Delineation of seed collection zones based on environmental and genetic characteristics for Quercus suber L. in Sardinia, Italy
iForest - Biogeosciences and Forestry, Volume 11, Issue 5, Pages 651-659 (2018)
doi: https://doi.org/10.3832/ifor2572-011
Published: Oct 04, 2018 - Copyright © 2018 SISEF
Research Articles
Collection/Special Issue: INCOTW - Sassari, Italy (2017)
International Congress on Cork Oak Trees and Woodlands
Guest Editors: Piermaria Corona, Sandro Dettori
Abstract
The assessment of seed zones or regions of provenance (RoP) to preserve local adaptation of tree species is an effective tool for the correct management of forest reproductive materials. The RoP for a species or sub-species is the area or group of areas subject to sufficiently uniform ecological conditions in which stands or seed sources show similar phenotypic or genetic characters, taking into account altitudinal boundaries where appropriate. However, the delineation of RoPs is commonly based on estimates of intrinsic environmental homogeneity, mainly climate and/or soil characteristics. The integration of genetic data into RoP maps is an important strategy to obtain a sound tool for managing forest reproductive materials. A study on Quercus suber (cork oak) in Sardinia (Italy) was carried out with the aim of determining ecological regions of provenance, investigating the genetic diversity among populations at the regional scale and identifying possible areas of interest for valorising the available germplasm. Identification of these areas was performed by Reserve Selection Analysis, which allows to identify priority areas by assessing the minimum number of sites required to include all the genetic diversity estimated by genetic analysis. Four spatial clusters were obtained based on environmental data: the northern and northern-eastern parts of the island were included in the Northern RoP; the second RoP covered the western part; and the third RoP enclosed the south-eastern region. The last group was distributed on the central part of the island (Central RoP) and includes the higher elevations. The sampled populations showed a low differentiation among populations and low diversity. According to the Reserve Selection Analysis, four conservation priority areas were identified. These indications can be useful at the local level because these sites can be proposed as stands for seed collection for future plantations.
Keywords
Regions of Provenance, Quercus suber, Seed Collection Zones, Spatial Genetic Structure, Sardinia
Introduction
The assessment of seed zones or regions of provenance (RoP) to preserve local adaptation of tree species is an important element for the correct management of forest reproductive materials (FRM), as well as for common practices in forestry and restoration activities ([72]). “Local-is-best” is the rationale underlying the delineation of tree seed zones ([15]), whose populations are expected to share similar environmental and climatic pressures; thus, planting populations adapted to local conditions allows both to preserve genotypes that are adapted to local conditions and to avoid the introduction of poorly adapted genotypes, which may cause loss of adaptive potential ([35]). In this perspective, local populations can ensure higher fitness in terms of survival, reproduction, productivity, disease resistance and abiotic resilience, than non-local materials ([35], [15]), unless climatic conditions change drastically.
In European countries, the identification of RoPs is regulated by the European Union (EU) directive 1999/105/CE and is a mandatory tool for the management of FRM. According to the directive, the RoP for a species or sub-species is the area or group of areas subject to sufficiently uniform ecological conditions in which stands or seed sources show similar phenotypic or genetic characters, taking into account altitudinal boundaries where appropriate. In such a context, two main approaches can be generally adopted, both based on clustering methods. When genetic data are available, a hierarchical clustering on sample populations may be performed ([39]) to aggregate stands sharing similar genetic traits. Although such a method represents the most accurate and reliable, genetic data are generally hard to be obtained. Moreover, an adequate sampling scheme would be highly expensive and difficult to be implemented for wide areas. For this reason, climate (i.e., ecological or environmental variability) is generally considered a good proxy for genetic variability. Actually, it is well known that climate mainly represents the most important driver for spatial distribution of forest species and reliable time series are one of the most relevant data for forest monitoring ([22]). In this case, the delineation of RoPs is commonly based on overall estimates of environmental homogeneity, mainly climate and/or soil characteristics ([49], [18]), and on a spatial clustering procedure. Main variables can be represented by long-term (30 years at least) averages of temperature and precipitation, and/or derived climatic indices which combine their values (e.g., aridity, continentality, etc. - [47]). However, the lack of species-specific genetic information in establishing RoPs could lead to underestimate the area of species range showing significant genetic differentiations, which may potentially cause increasing levels of inbreeding among planted trees ([41]). Therefore, integrating RoP maps with genetic data is an effective strategy to obtain a sound tool for the legislation of FRMs.
The climatic context of the Sardinia island has often been described in the literature as an example of a thoroughly Mediterranean island. Recently, Canu et al. ([6]) have classified the different environments across the island in 43 ecological groups, according to well know climatic belts. However, such groups are too many for a viable and cost-effective management of Forest Genetic Resources under the 1999/105/CE directive. For this reason, in 2016 the Regional Government of the Autonomous Region of Sardinia (Italy) developed a program aimed to screen seed sources and delimit RoPs with a specific target on cork oak (Quercus suber L.) populations, in order to protect and valorise available gene pools. Cork oak is a widely distributed species across the Mediterranean basin, both on the European side (Italy, France, Spain, Portugal) and North Africa (Tunisia, Morocco, Algeria). The species is widely cultivated in the Iberian Peninsula (Portugal, Western Spain, Catalonia and Balearic Islands) and France (Provence, Corsica, Aquitaine, Languedoc). In Italy, it is present along the Tyrrhenian coastline (Tuscany and Latium), on the main islands (Sicily and Sardinia), on the ionic Calabrian side and in Apulia. Cork oak is generally sporadic, and best growing in areas with cold, moist winters and hot summers. It is commonly found at 300-600 m but can occur up to 1000 m a.s.l. It prefers acid soils and avoids limestone and carbonated substrates. Sardinian cork oak stands cover about 140.000 ha ([32]) and have great socio-economic importance in the region due to its exploitation for cork extraction and processing, and for silvopastoral activities ([9], [61]). However, the risk of losing cork forests is high because of forest fires and diseases, overgrazing, land abandonment and exploitation, with subsequent biodiversity loss and deterioration of ecosystem services ([11]), exacerbated by the projected climate changes ([23]).
Studies of the genetic and geographical structure of Q. suber have been previously reported on different populations around the Mediterranean range using different molecular markers ([44]). First molecular characterization of Sardinian germplasm was accomplished by RAPD markers in several economically and agriculturally strategic stands ([5]). However, to establish a detailed RoP map for the species, an updated and refined assessment of the genetic structure of Sardinian populations is required. To this purpose, microsatellite markers represent a valid method for the assessment of genetic diversity due to their high levels of polymorphism, their codominant nature and their transferability.
The objectives of this study are: (1) to determine the ecological regions of provenance for Cork oak in Sardinia; (2) to investigate the genetic diversity among Cork oak populations at the regional scale; (3) to identify possible areas of interest to be selected as seed stands.
Materials and methods
RoP delineation based on environmental variables
An ensemble of current climate data and soil characteristics was used to analyse the environmental variability across Sardinia. The ClimateEU software (freely available at ⇒ https://sites.ualberta.ca/~ahamann/data/climateeu.html) is a standalone software which can be used to downscale 4-km raster surfaces at higher spatial resolutions according to local elevation. Parameter-elevation Regressions on Independent Slopes Model (PRISM) monthly data (0.5° resolution) can be extracted and downscaled to scale-free point data ([14]), and the value of seasonal and annual climate variables for specific locations can be calculated based on latitude, longitude and elevation. The original meteorological data is derived from the CRU-TS 3.22 database ([26]), which includes more than 2000 meteorological stations across the whole Europe and 13 stations in the region of interest (within the grid box centred at N37.5-E7.5 and N42.5-E7.5). Such dataset has proven to be highly suitable for local studies in the Mediterranean region even when long time series are required, thanks to the fair representativeness of local trends ([46]). Variable surfaces are generated using lapse-rate adjustments according to latitude and longitude ([70]), based on the difference between the grid elevation and the elevation of the location of interest.
The software was used to generate high-resolution (250 m) raster maps of 36 biologically-relevant climatic variables (such as growing and chilling degree days, heating and cooling degree days, Hargreaves moisture deficit and reference evaporation, etc. - see Tab. S1 in Supplementary material) for the 1981-2010 normal period in Sardinia. A routine was created by calculating the downscaled values for each pixel. In addition, soil data (pedology and soil characteristics) were derived from the Italian eco-pedologic map ([12]).
All the obtained maps were then geo-referenced in the ETRS89/UTM32N (EPSG: 25832) reference system and used to perform a spatial principal component analysis (sPCA). All the data were centred and scaled separately. A standardisation procedure was applied as the variables were expressed in different scales (e.g., degrees Celsius for temperatures, millimetres for precipitation, pure numbers for indices). The aim of sPCA was to include as much environmental variability information as possible in further analysis (clustering), avoiding at the same time the collinearity between variables. As PCA compresses raw data variability into few uncorrelated (new) variables, PC components are not affected by collinearity between environmental factors. Only a subset of PC components was used and subjected to unsupervised classification ([27]) summing up to 99% of the explained variance.
Hierarchical clustering was applied to PC data in a GIS environment (“i.cluster” and “i.maxlik” functions of GRASS-GIS - [24]) using as weight the eigenvector of each ecological variable for each component obtained from PCA. Pairwise Euclidean distances between pixels were calculated for each variable. The maximum number of iterations was set to 50, while the convergence value (the points at which cluster means become stable) was set to 98% ([47]). The number of spatial clusters was set based on a dendrogram computed using the PC scores of 1000 points randomly distributed across Sardinia, regardless Quercus suber distribution.
Plant material and DNA extraction
A total of 277 individuals were randomly sampled at 10 sites (Tab. 1) for genetic analyses. Leaves were collected from individual trees and kept under silica gel for long-term conservation. Total genomic DNA was extracted by grinding 50-60 mg of dry material in liquid nitrogen and using the DNeasy® Plant mini kit (QIAGEN, Hilden, Germany). The extracted DNA was diluted to 5 ng µl-1 of working solution and quantified by spectrophotometry (Eppendorf Biophotometer® D30, Eppendorf AG, Hamburg, Germany).
Tab. 1 - Main environmental characteristics of the sampling sites. (Pop): population label; (Site): official forest name; (#ind): number of sampled individuals; (N): latitude (°); (E) longitude (°); (Alt): altitude (m a.s.l.); (MAT): mean annual temperature (°C); (MAP): mean annual precipitation (mm); (AHM): annual heat:moisture index (MAT+10)/(MAP/1000); (RoP): region of provenance each population belongs to. Further details of climate for each site are reported in Tab. S2 (Supplementary material).
| Pop | Site | #ind | N | E | Alt | MAT | MAP | AHM | RoP |
|---|---|---|---|---|---|---|---|---|---|
| LIM | Limbara Sud | 24 | 40.81 | 9.19 | 457 | 15.0 | 671 | 37 | 1 |
| MOL | Monte Olia | 28 | 40.75 | 9.31 | 486 | 14.1 | 679 | 36 | 1 |
| TER | Terranova | 26 | 40.68 | 9.38 | 594 | 14.0 | 670 | 36 | 1 |
| LIT | Sos Littos | 30 | 40.64 | 9.48 | 145 | 16.3 | 545 | 48 | 3 |
| FIO | Fiorentini | 25 | 40.52 | 9.05 | 799 | 13.1 | 827 | 28 | 4 |
| BAR | Barigadu | 29 | 40.08 | 9.02 | 649 | 13.9 | 785 | 30 | 4 |
| MAN | Monti Mannu | 19 | 39.38 | 8.67 | 558 | 14.3 | 708 | 34 | 2 |
| MAR | Marganai | 19 | 39.35 | 8.57 | 686 | 13.7 | 751 | 32 | 2 |
| SET | Settefratelli | 34 | 39.31 | 9.37 | 451 | 15.2 | 466 | 54 | 3 |
| SUL | Gutturu Mannu | 43 | 39.10 | 8.88 | 328 | 15.5 | 604 | 45 | 2 |
Microsatellite analysis
Samples were analysed with a set of 5 polymorphic nuclear markers (nuSSR - Tab. 2), previously developed for other oak species (QpZAG9, QpZAG15, QpZAG36, QpZAG110 in Q. petrea (Matt) Liebl.; QrZAG7 in Q. robur L.). As a rule, SSRs are species-specific markers, which must be developed de novo for each species, mainly because they usually occur in non-coding regions of the genome, which are not highly conserved. However, Mendelian inheritance and transferability of nuSSRs from other oak species to cork oak had been previously demonstrated ([30], [62]). The initial set of markers also included MSQ4 ([17]), QrZAG20 ([37]) and QpZAG46 ([63]), though they have been subsequently discarded for poor/none amplification and/or unreliable scoring in most samples.
Tab. 2 - nuSSR markers selected for molecular analysis. (Na): number of alleles; (Ne): effective numbers of alleles; (He): expected heterozigosity; (Fis): fixation index; (ƒna): null allele frequency.
The five markers selected in this study showed a pattern of allelic richness and diversity similar to that obtained using three to seven nuSSRs markers in cork oak by Löpez-Aljorna et al. ([42]), Da Costa ([13]) and Zucca ([73]). The latter author analysed three populations from northern, central and southern Sardinia using seven nuSSRs, of which five are in common with our study. Comparing the diversity at each locus with our results (see below), we found a double amount of alleles (22 versus 11) at locus ZAG110, while at the other loci (ZAG36, ZAG9, ZAG15 and ZAG7) the number of alleles was similar, ranging between 4 and 10. Thus, we selected a posteriori the five markers best discriminating among individuals and populations. Moreover, the unbiased probability of identity (uPI - [51]) was computed to detect the chance that two individuals drawn at random from a sample will have the same genotype at multiple loci. The estimated uPI for the combination of the five selected markers over all populations was 1.3 × 10-4.
PCRs were performed in triplexes in Eppendorf Mastercycler® pro (Eppendorf AG, Hamburg, Germany). Each sample was amplified in a 12.5 µl total volume reaction according to QIAGEN Type-it® Microsatellite PCR kit (QIAGEN, Hilden, Germany). Cycling protocol was as follows: 95 °C for 5 min; 28 cycles at 95 °C for 30 sec, 57 °C for 90 sec, 72 °C for 30 sec; 60 °C for 30 min. 1 µl of each amplification product was added to a mixture of 20 µl HIDI formamide and 0.3 µl LIZ and denatured at 95 °C for 5 min. Samples were run on ABI Prism 3130® Avant DNA sequencer (Applied Biosystems, Foster City, CA, USA). Results were analysed by GeneMapper® Software (Life Technologies, Carlsbad, CA, USA), and allelic profiles were scored by automatic binning and visual checking.
Statistical analysis
Number of observed (Na) and effective (Ne) alleles, observed (Ho) and expected (He) heterozygosity ([50]) were calculated using GENALEX 6.5 software ([52], [53]). The inbreeding coefficient (Fis) for each population and each locus was obtained computing a hierarchical AMOVA using the software ARLEQUIN ver. 3.5.2.2. Statistical significance was determined by a non-parametric approach using 1000 permutations ([21]). The assessment of allelic richness by measuring allele frequencies must take into account the variation in population sizes (Tab. 1). For this reason, allelic richness (A) and the number of private alleles (Pa) were computed using the rarefaction method with the HP-RARE software ([36]). Null allele frequency for each locus was estimated using the package FreeNA ([7]). Principal coordinates analysis (PCoA) was performed with GENALEX 6.5 using a pairwise population matrix of Nei’s unbiased genetic distance ([50]). Correlations among sites for measures of genetic, environmental and geographic distance were calculated by Mantel test and partial Mantel tests using the software PAST ver. 3.19 ([25]), with 9999 permutations. Partial Mantel tests were conducted to determine the strength of the correlation between two distance matrices aimed at assessing the dependence between two matrices of distances while controlling the effect of a third distance matrix.
Genetic structure was evaluated by a Bayesian approach performed using STRUCTURE software ver. 2.3.4 ([55]). Twenty iterations for each value of K between 1 and 10 were run with a burning period of 10.000 and 105 Markov chain Monte Carlo cycles. The most likely number of clusters was determined by the Evanno’s method ([20]), using the STRUCTURE HARVESTER software ([19]). Twenty runs were averaged by CLUMPP ver. 1.1.2 web service ([33]) and graphically represented using the software DISTRUCT ver. 1.1 ([58]).
Selection of priority areas and overlay with regions of provenance
Grids for genetic parameters were generated in DIVA-GIS software ver. 7.5 ([28]), based on a grid cell of 6 minutes and applying a circular neighbourhood with a diameter of one degree. Four different classes of alleles can be defined: (i) rare alleles with local distribution (private); (ii) frequent alleles observed with local distribution; (iii) rare alleles observed in a large area; (iv) frequent alleles commonly distributed. Priority for conservation should be given to populations that retain frequent alleles with local distribution, indicating the presence of genotypes adapted to specific environments. However, the Reserve Selection method implemented in DIVA-GIS and developed by Rebelo & Siegfried ([57]) considered all these classes for the identification of the priority conservation areas. A point-to-grid analysis was applied, selecting the “Complementary” option and giving equal weight to each allele. This method allows the selection of conservation priority areas not only based on their allelic richness but also on differences/complementarity in allelic composition, by identifying the minimum number of sites needed to include most of the genetic diversity estimated by nuclear markers. Then, the procedure ranks the geographic units by indicating the first chosen area as that with the highest allelic richness; each successive area selected best aggregates the intra-specific diversity already represented within the previously selected priority area.
Finally, the selected zones (seed stand candidates) were overlaid onto the RoPs as characterised above in the same GIS environment.
Results
Regions of provenance
Four groups were selected by hierarchical clustering as best matching between environmental variability and spatial fragmentation. In the sPCA, 99% of the cumulated variance was accounted for by five components, which were selected as input for the GRASS-GIS modules. As a result, four spatial clusters were obtained (Fig. 1). The northern and northern-eastern part of the island was included in a unique RoP, named Northern Region, RoP1. The western and south-eastern parts were included in RoP2 and RoP3, respectively. The last group mainly included the higher elevation areas in the central part of the island (Central region, RoP4). RoP1 and RoP4 were colder than RoP2 and RoP3 concerning mean annual temperature (MAT); RoP1 was the driest region and RoP4 the wettest. The average climatic values and extremes of each RoP are reported in Tab. S2 (Supplementary material).
Fig. 1 - The regions of provenance (RoP) obtained in this study based on climatic variables.

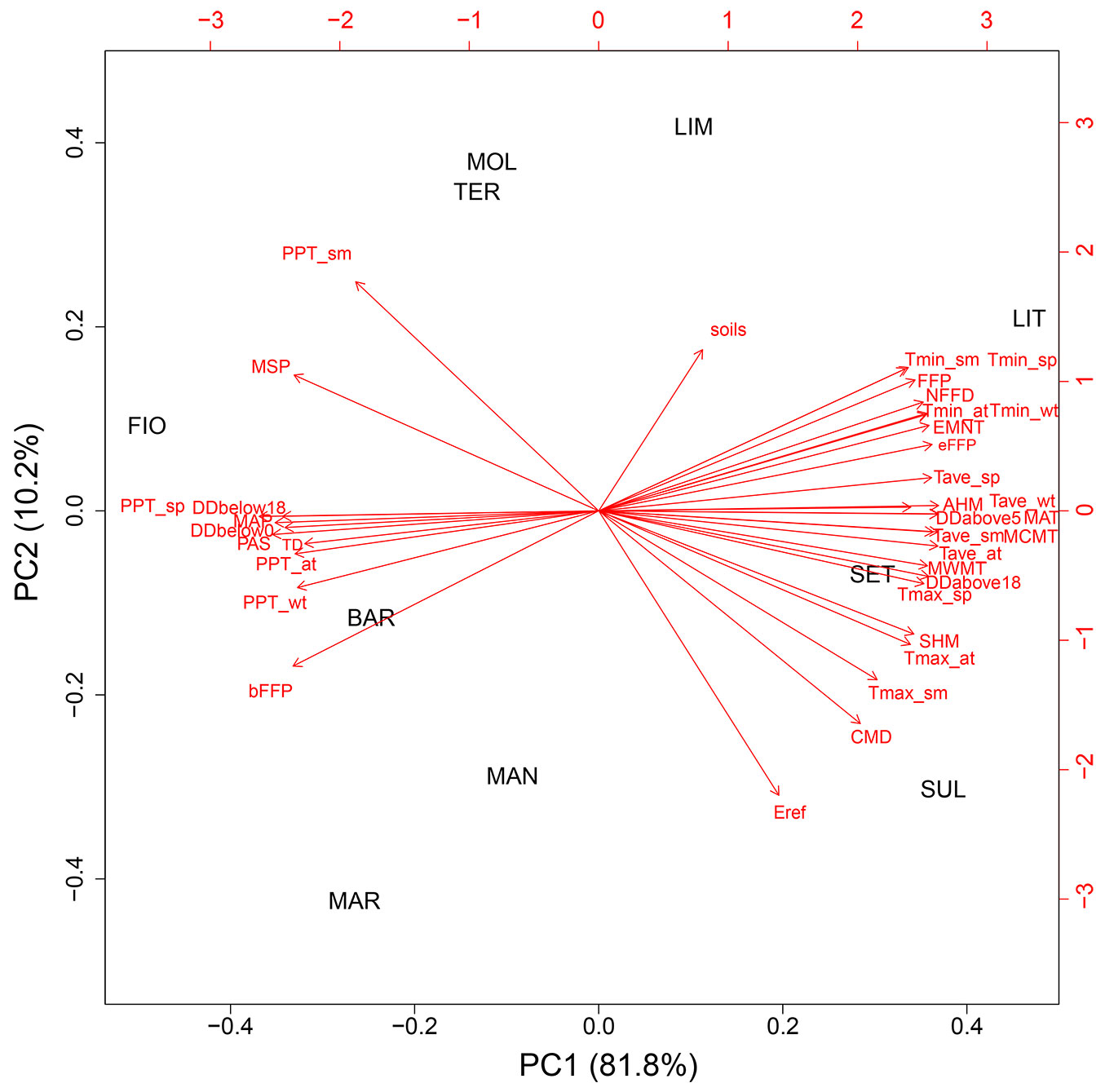
Concerning the climate characteristics of sampling sites, the average values of the 36 climatic variables considered are reported in Tab. S3. Two main clusters of sampling sites were detected by multivariate analysis (PCA) based on the selected 36 climatic variables (Fig. 2). One cluster grouped the southern sites SUL, SET, MAN and MAR, including the northern LIT, which also has the lowest altitude; the other cluster grouped the northern sites MOL, LIM, TER, FIO and BAR mainly according to the directions of the minimum temperatures, soils, the beginning of the frost-free period and the variables related to seasonal precipitation in autumn and winter. Accordingly, a good correlation between geographic and climatic distance matrices was detected (r = 0.45, p < 0.05).
Fig. 2 - Principal component analysis (PCA) of the sampling sites according to climatic variables.
Genetic diversity
In Tab. 2 genetic diversity parameters for each locus are reported. The number of alleles per locus (Na) ranged from 2.7 for locus QpZAG9 to 13.9 for QpZAG110. The highest number of effective alleles (Ne) was observed at locus QpZAG110. Expected heterozygosity (He) was higher for QpZAG110 (0.88) than the other analysed loci. Fixation index (Fis) showed no significant departure from the Hardy-Weinberg equilibrium. Null allele frequency ranged between 0 and 0.043 (Tab. 2). Global Fst for all loci and pairwise Fst were very similar when estimated from raw allele frequencies or allele frequencies corrected for null alleles (data not shown). Thus, null alleles had very little influence on this analysis, and all further tests were performed using uncorrected allele frequencies.
Tab. 3 reports the genetic diversity parameters estimated at each sampling site. The number of alleles (Na) ranged from 37 (SUL) to 28 (MAR). Allelic richness (A) showed values from 6.3 (SUL and TER) to 5.6 (MAR). The number of private alleles ranged from 0.0 (LIT, FIO, BAR, MAR) to 0.3 (SUL). Expected heterozygosity (He) showed homogeneity among the populations, varying around 0.56. Fixation indices (Fis) showed no significant departure from zero in all populations and varied between -0.03 (MAN) to 0.07 (LIM).
Tab. 3 - Genetic diversity parameters estimated at each sampling site. (Na): total number of alleles; (A): allelic richness; (Pa): number of private alleles; (He): expected heterozigosity; (Fis): fixation index.
| Pop | Na | A | Pa | He | Fis |
|---|---|---|---|---|---|
| LIM | 32 | 6.01 | 0.22 | 0.52 | 0.069 |
| MOL | 33 | 5.91 | 0.12 | 0.52 | -0.015 |
| TER | 35 | 6.30 | 0.18 | 0.55 | -0.015 |
| LIT | 35 | 6.27 | 0.01 | 0.58 | 0.063 |
| FIO | 31 | 5.95 | 0.02 | 0.57 | -0.040 |
| BAR | 32 | 5.91 | 0.01 | 0.56 | 0.046 |
| MAN | 32 | 6.40 | 0.12 | 0.60 | -0.034 |
| MAR | 28 | 5.60 | 0.01 | 0.57 | -0.040 |
| SET | 34 | 5.91 | 0.16 | 0.58 | 0.057 |
| SUL | 37 | 6.31 | 0.33 | 0.56 | -0.005 |
AMOVA analysis (Tab. 4) showed a low differentiation among populations (Fst = 0.009, p = 0.49), not significantly different from 0. The molecular variance among individuals within populations was Fis = 0.024. Fis was not significantly different from 0 (p = 0.06). Molecular variance was therefore mainly located at the individual level (Fit = 0.033, p < 0.05).
Tab. 4 - Results of the analysis of molecular variance (AMOVA).
| Source of variation |
df | Variance components |
% variation |
F-stats | Value | Prob. |
|---|---|---|---|---|---|---|
| Among populations | 9 | 0.013 | 0.94 | Fst | 0.009 | 0.49 |
| Among individuals w/ populations | 267 | 0.034 | 2.41 | Fis | 0.024 | 0.06 |
| Within individuals | 277 | 1.37 | 96.65 | Fit | 0.033 | 0.020 |
| Total | 553 | 1.41 | 100.00 | - | - | - |
Genetic structure
Nei’s unbiased genetic distance (uNei) values among populations varied between 0.001 and 0.059 (Tab. 5). Significant differences were observed among MAR and all the other populations. SET and SUL showed some significant distances with northern and central populations. Also the distance between SET and SUL, the southernmost populations, was significant.
Tab. 5 - Pairwise population matrix of Nei’s unbiased genetic distance. (*): p<0.05; (**): p<0.01; (***): p<0.001.
| - | LIM | MOL | TER | LIT | FIO | BAR | MAN | MAR | SET |
|---|---|---|---|---|---|---|---|---|---|
| MOL | 0.001 | 0 | - | - | - | - | - | - | - |
| TER | 0.001 | 0.000 | 0 | - | - | - | - | - | - |
| LIT | 0.010 | 0.009 | 0.000 | 0 | - | - | - | - | - |
| FIO | 0.004 | 0.005 | 0.000 | 0.000 | 0 | - | - | - | - |
| BAR | 0.004 | 0.013 | 0.000 | 0.001 | 0.000 | 0 | - | - | - |
| MAN | 0.009 | 0.000 | 0.010 | 0.007 | 0.005 | 0.013 | 0 | - | - |
| MAR | 0.029* | 0.030** | 0.025** | 0.023* | 0.027* | 0.037** | 0.038* | 0 | - |
| SET | 0.042*** | 0.024*** | 0.015** | 0.000 | 0.013 | 0.024*** | 0.012 | 0.050*** | 0 |
| SUL | 0.010 | 0.016* | 0.014* | 0.001 | 0.009 | 0.013* | 0.001 | 0.059*** | 0.019*** |
Mantel test between genetic and the geographic distances (IBD = isolation by distance) showed a significant correlation coefficient (r = 0.23, p < 0.05). Differently, the correlation between genetic and climatic distances was not significant (r = 0.18, p = 0.12). These results were confirmed by the partial Mantel test: when geographical factors were controlled a lack of isolation by climatic distance (r = 0.16, p = 0.18) was observed; whereas when climatic factors were controlled, we detected significant correlations between genetic differentiation and geographical distance (r = 0.32, p < 0.05).
The analysis of population structure revealed that all populations belonged to the same deme (data not shown).
Selection of priority populations and overlay with the regions of provenance
According to the Reserve Selection Analysis, four priority areas could be delimited (Tab. 6). Priority 1 was assigned to TER in the RoP1 Northern. Priority 2 was located in SUL in the RoP2 Western. An area of Priority 3 was located in SET, in RoP3. The LIM population was reported as an area of Priority 4, located in the RoP1. These sites contained almost the entire set of alleles detected in the island in this survey, missing only one allele (ZAG9_234) out of 52, present in only two sites (MAR and MAN) with a frequency around 5%. At the opposite, the four private alleles (measured only in one site with a frequency averaging 3%) were comprised in the selected sites (Fig. S2 in Supplementary material).
Tab. 6 - Priority areas identified by Reserve Selection Analysis. The number in parentheses refers to the corresponding region of provenance (RoP).
| Priority area |
Population name (RoP) |
|---|---|
| 1 | TER (1) |
| 2 | SUL (2) |
| 3 | SET (3) |
| 4 | LIM (1) |
Discussion
Environmental features, such as climate and soil characteristics, play important roles in determining tree population dynamics. Thus, the delimitation of RoPs is mainly based on the spatial homogeneity of environmental parameters. Their number and size vary greatly among countries, due to different approaches and criteria used for their delineation, based on ecological units or vegetation zones, phenotypic or genetic similarities (or their combination), the economic importance of the species ([40]). In some countries, including Italy, RoP delineation is done not at the national but at the regional level. Concerning the whole Sardinian environment, four regions were detected, which minimised the ecological variability within and maximised differences among RoPs. Although previous authors identified a higher number of environmental groups as adequate to explain and characterize the whole ecological variability of the island ([6]), such studies were always referred to more general spatial analyses (i.e., pure climatological classifications), aiming at including Sardinia in a broad European context and matching the local climate with the European ones. Indeed, a high number of groups is often unsuitable for cost-effective and straightforward management of FGR at the regional level. In fact, RoPs delineation should take into account the trade-off between statistical accuracy and practical complexity ([40]), in view of seeds and FRM transferability ([67]).
Regarding the species-specific RoPs for cork oaks, Varela ([68]) proposed seven RoPs for Portugal, based on environmental parameters, such as climate, lithology and management. Similarly to other studies for different tree species ([34], [3], [65]), these regions usually overlap with well-delimited areas easily identified for practical operations. In this study, a different approach strictly based on spatialised ecological parameters was used, also taking into account elevation. As a consequence, our map shows numerous small enclosed areas of RoP4, laying on the more elevated portion of the island, surrounded by areas belonging to a different RoP.
As expected, the populations sampled in this study revealed a low genetic differentiation and diversity. Although the seed dispersal is too low to be a determining factor in homogenising oak populations, the high level of pollen flow could influence variability within populations and contribute to decrease the inter-population variability ([42]), similarly to other species with an extensive gene flow and long distance anemophilous pollination ([45], [54], [2]). In addition, in the case of cork oak, anthropogenic pressure cannot be excluded, considering the long (for at least 2000 years) exploitation of the species with likely intentional acorn transport ([64]).
Molecular characterisation of Sardinian cork oak populations has already been assessed through RAPD markers ([5]), whose results revealed a significant differentiation among populations, differently from our study. Although the sampled populations do not coincide in these two studies, a different pattern could be expected, as RAPD markers exhibit a certain lack of reproducibility due to mismatch annealing and competition for amplification among various fragments ([16], [38]). More recently, Zucca ([73]) analysed different Mediterranean cork oak provenances in a common garden by nuSSR, confirming the low diversity of Sardinian populations compared to other European provenances (on average lower by 30%) and a concurrent low differentiation. Likewise, a low differentiation among populations and diversity within population was shown by AFLP markers among Portuguese cork oaks ([8]).
Based on the lack of well-defined different gene pools, seed collection should be sampled regardless of the location. However, through Reserve Selection Analysis, we determined the minimum number of areas to be sampled for including the highest number of alleles and private/rare alleles. According to Hoban & Schlarbaum ([29]), the goal of capturing all alleles with high probability may be unrealistic, while a reasonable goal of sampling many but not all alleles in the various categories (local rare, local common, etc.) may be preferred. This could help identify key sites for seed collection activities, because resources allocated for these activities are frequently scarce, limiting the possibility to conduct massive samplings in several areas. According to the Reserve Selection Analysis carried out, the areas of high priority identified in this study hold the majority of allelic richness and private alleles for cork oak in Sardinia: each selected population complemented at best the intra-specific diversity already represented within the previously selected priority area. As a consequence, the second-ranking priority area was not necessarily that with the second highest diversity (given that a large portion of this diversity had already been enclosed in the first priority area), but rather areas showing the allelic composition best complementing to that of the already prioritised area. Indeed, the four selected sites for cork oak in this study were not those with the highest rank of allelic richness, nor the ones with the highest rank of private alleles. The four priority areas were recognised in the northern sites TER and LIM and southern sites SET and SUL, mainly following the distribution of common alleles at the local scale and selecting almost the whole allelic pattern, without missing any private alleles. The sites covered three out of four RoPs, while no population was selected in the high elevation RoP4. Reserve Selection Analysis has widely been applied at broad (continental) scale, for selecting in-situ conservation areas for endangered species ([66], [69]), but also for defining seed collection zones ([31]). Our findings support previous suggestions about ex-situ conservation and seed collection, confirming that material should come from geographically or ecologically distant populations to maximise the collection of genetic diversity ([71]).
The selection of spatially distributed populations may also better represent the existing quantitative variation across the species range. In fact, it is known that neutral molecular diversity of populations does not always reflect their actual variation of quantitative traits. On the other hand, it has been reported that many plant populations are adapted to their local habitats, i.e., populations often include genetic variants showing a higher fitness in the local environment. Our study revealed a moderate isolation by distance (IBD, the correlation between genetic and geographic distances), although the correlation between genetic and climatic distance among sites was not significant. However, we could observe some significant genetic distances between several southern and northern populations. This suggests a likely influence of environmental conditions on the fitness of these cork oak populations, more related to latitude than altitude. The hypothesis is supported by Ramírez-Valiente et al. ([56]), who asserted that, in spite of the low genetic diversity and the weak among-population divergence using molecular markers, cork oak was able to develop local adaptations in response to natural selection. Additionally, in cork oak seedlings exposed to high irradiance and drought under controlled environment, provenances from dry places exhibited higher tolerance to these environmental stressors, despite no relevant change in morphology ([1], [59]). Other studies analyzed the variability of candidate genes related to different environmental conditions, suggesting the existence of local adaptations ([48], [43]). Epigenetic regulation has also been reported as an important factor for adaptation of cork oak to specific environmental stresses ([10]).
Therefore, in order to improve this first assessment on the genetic resources of cork oak in Sardinia, common gardens and transplanting along the island in different RoPs will be necessary ([4]): these would provide tools to characterise provenances according to their genetic differentiation and plastic responses ([60]), especially to drought, the main threat in the Mediterranean region.
Conclusions
This study integrated the information on the population genetic structure of cork oak by nuSSR markers with environmental characteristics of Sardinia in order to delineate regions of provenance for the species. Four RoPs were selected as best matching between environmental variability and spatial fragmentation. Almost the entire genetic diversity was observed at the individual level. According to the Reserve Selection Analysis, four priority areas were identified which are proposed as seed collection sites for cork oak in Sardinia. These findings can be useful for future management of cork oak genetic resources and future plantations in the island. However, further efforts aimed to investigate the functional response of different provenances are necessary by common gardens and provenance trials along the island.
Acknowledgements
This study was funded by the Project LR 7/2007 TENDER “Multifunzionalità delle foreste a quercia da sughero” - Regione Autonoma della Sardegna. The authors want to thank the Agenzia Fo.Re.S.T.A.S., in particular, Gen. Manager dr. Casula, Dr. Sara Maltoni, forest officers, workers. Molecular analyses by sequencer were performed at the National Research Council - Institute of Agro-environmental and Forest Biology (CNR-IBAF) in Porano (TR, Italy), assisted by Marcello Cherubini.
References
CrossRef | Gscholar
Online | Gscholar
Gscholar
Gscholar
Online | Gscholar
CrossRef | Gscholar
CrossRef | Gscholar
Gscholar
Gscholar
Authors’ Info
Authors’ Affiliation
Angela Teani
Maurizio Marchi
Maria Cristina Monteverdi
Fulvio Ducci
Council for Agricultural Research and Economics, Research Centre for Forestry and Wood, v.le Santa Margherita 80, I-52100 Arezzo (Italy)
National Research Council, Institute of Agro-environmental and Forest Biology (CNR-IBAF), v.le Marconi, Porano, TR (Italy)
Corresponding author
Paper Info
Citation
De Dato G, Teani A, Mattioni C, Marchi M, Monteverdi MC, Ducci F (2018). Delineation of seed collection zones based on environmental and genetic characteristics for Quercus suber L. in Sardinia, Italy. iForest 11: 651-659. - doi: 10.3832/ifor2572-011
Academic Editor
Piermaria Corona
Paper history
Received: Jul 29, 2017
Accepted: Aug 02, 2018
First online: Oct 04, 2018
Publication Date: Oct 31, 2018
Publication Time: 2.10 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2018
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 50127
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 40517
Abstract Page Views: 4367
PDF Downloads: 4087
Citation/Reference Downloads: 3
XML Downloads: 1153
Web Metrics
Days since publication: 2861
Overall contacts: 50127
Avg. contacts per week: 122.65
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Jul 2026)
Total number of cites (since 2018): 10
Average cites per year: 1.11
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Fine-scale spatial genetic structure in a multi-oak-species (Quercus spp.) forest
vol. 8, pp. 324-332 (online: 05 September 2014)
Technical Reports
Conservation and use of elm genetic resources in France: results and perspectives
vol. 13, pp. 41-47 (online: 03 February 2020)
Review Papers
Genetic diversity and forest reproductive material - from seed source selection to planting
vol. 9, pp. 801-812 (online: 13 June 2016)
Editorials
Workshop COST E52 “Evaluation of beech genetic resources for sustainable forestry”
vol. 2, pp. 104 (online: 10 June 2009)
Research Articles
Growth, spring phenology and stem quality of four broadleaved species assessed in provenance trials in the Netherlands - Implications for seed sourcing
vol. 18, pp. 242-251 (online: 22 September 2025)
Research Articles
Networking sampling of Araucaria araucana (Mol.) K. Koch in Chile and the bordering zone of Argentina: implications for the genetic resources and the sustainable management
vol. 2, pp. 207-212 (online: 22 December 2009)
Commentaries & Perspectives
The genetic consequences of habitat fragmentation: the case of forests
vol. 2, pp. 75-76 (online: 10 June 2009)
Technical Reports
Population genetic structure of Platanus orientalis L. in Bulgaria
vol. 4, pp. 186-189 (online: 11 August 2011)
Research Articles
Seedling emergence capacity and morphological traits are under strong genetic control in the resin tree Pinus oocarpa
vol. 17, pp. 245-251 (online: 16 August 2024)
Research Articles
The maternal environment of European beech (Fagus sylvatica L.) affects intrapopulation variability in seed traits and germination
vol. 19, pp. 114-121 (online: 10 April 2026)
iForest Database Search
Search By Author
Search By Keyword
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords
PubMed Search
Search By Author
Search By Keyword







