Clonal structure and high genetic diversity at peripheral populations of Sorbus torminalis (L.) Crantz.
iForest - Biogeosciences and Forestry, Volume 9, Issue 6, Pages 892-900 (2016)
doi: https://doi.org/10.3832/ifor1885-009
Published: May 29, 2016 - Copyright © 2016 SISEF
Research Articles
Abstract
Knowing the level of genetic diversity and structure in marginal plant populations is essential for managing their genetic resources. This is particularly important for rare scattered tree species, such as Sorbus torminalis (L.) Crantz. We investigated the genetic diversity and its spatial distribution in peripheral populations of S. torminalis. As the species is known to reproduce vegetatively, we also evaluated clonal structure within populations. Using 13 nuclear microsatellite loci designed in two multiplexes, we genotyped 172 individuals revealing the existence of 100 distinct genotypes. Number of ramets per genotype was variable across populations with an average of 1.72. Examples of somaclonal variation at particular loci were detected. Measures of genetic diversity of the total sample were relatively high (mean allelic richness AR = 10.293; expected heterozygosity He = 0.756), as compared to other S. torminalis populations. We noticed a slightly negative inbreeding coefficient (FIS = -0.029) indicating a small excess of heterozygotes, which is typical for self-incompatible plants. Genetic differentiation among populations was low (FST = 0.048), but Bayesian clustering methods revealed the existence of three distinct genetic clusters only in part related to population structure. Significant spatial genetic structure within populations was also detected (Sp = 0.0125) indicating fine-scale pattern of isolation by distance. Our study demonstrated that peripheral populations of S. torminalis may exhibit relatively high levels of genetic diversity despite the existence of vegetative propagation. Nevertheless, if the studied or similar populations are expected to be utilized as seed sources for ex-situ or in-situ conservation purposes, the existence of clonal structure and spatial genetic structure must be taken into account in order to avoid excessive sampling of the same or closely related genets.
Keywords
Sorbus torminalis, Clonality, Range Limits, Somatic Mutations, Microsatellites
Introduction
Genetic diversity is considered one of the major determinants assuring long term stability of plant populations. Loss of genetic diversity is expected to decrease population fitness and may become a limiting factor when populations face unpredicted environmental changes ([21], [39]). However, the distribution of genetic diversity within the range of a species is often not uniform, with genetic diversity usually decreasing towards the limits of the species distribution ([20]). This is mostly due to the increased fragmentation of populations towards range limits, coupled with restricted gene exchange among populations. Consequently, loss of genetic diversity is more likely in small isolated peripheral populations due to random genetic drift ([81]). Therefore, peripheral populations may evolve separately from the core populations ([26]) and may possess unique genetic variation due to local adaptation to marginal habitats. Their genetic pool might be important for maintaining the overall species capacity to evolve and adapt to changing environmental conditions ([47]).
Climate changes predicted in Europe are expected to affect a number of plant species, including forest trees ([44]). Although changing climatic conditions may cause the loss of existing habitats, there may also be opportunities for some species to colonize new areas where climate conditions will become more favorable ([34], [45]). Thus, knowledge of genetic diversity in peripheral populations may be useful in designing conservation strategies for a species and assessing potential for its further spread.
Forest management practices in Central Europe favor dense, even aged forests with closed-canopies. This causes the loss of habitats for many other early successional tree species of lower economical importance. One such example is the wild service tree (Sorbus torminalis [L.] Crantz), a typical forest companion tree species. The species occurs in small, isolated groups or as scattered individual trees ([3], [16], [35], [64]). It is distributed across western, central and south-western parts of Europe, but occurs also in northern Africa and in south-western Asia ([16]). The species reaches its north-eastern range limit in Poland, where it grows in small scattered populations, mainly in the western part of the country ([8]). In Poland the species is classified as endangered and protected by state law ([9]).
Wild service tree is pollinated by insects (hymenopteran or dipteran) and is capable of long distance pollen dispersal ([56], [57]). The species is characterized by gametophytic self-incompatibility system ([51]) suggesting a predominantly outcrossing mating system (but see [36]). Seeds are efficiently dispersed by birds and mammals, which promote the high colonizing abilities of the species ([57], [67], [63]). Nevertheless, sexual reproduction, especially in northern peripheral populations was found to be limited by short growing seasons and inbreeding depression resulting from low population sizes ([64], [66]). Although inbreeding depression coupled with small population sizes may cause populations to be vulnerable to extinction, wild service tree has the ability for clonal growth ([35], [66], [67]) which occurs primarily based on root sprouts ([65]). This may facilitate the persistence of populations that would otherwise be limited by sexual reproduction ([73]).
Although several studies report on the genetic diversity of S. torminalis populations throughout Europe ([15], [54], [35], [3], [11]), studies focusing on peripheral populations of the species are scarce ([66]). The latter authors found that small populations located at the northern limits were characterized by high clonality and low genetic variation ([66]).
In this paper we investigated a group of small, fragmented populations of the wild service tree growing at the north-eastern species distribution limits. Using nuclear microsatellite markers, we assess the extent of clonality and the levels of genetic diversity. We also investigated the genetic structure within and among the studied populations. The results are discussed in the context of the management of genetic resources of the species.
Materials and methods
Plant material sampling
We sampled plants of Sorbus torminalis throughout populations located in the Forest District Jamy (Poland), which is the north-eastern range limit of the species (18° 51′ - 19° 00′ N, 53° 29′ - 53° 35′ E). These populations have been previously described in detail by Bednorz ([8]), who documented 203 adult trees in this region. We attempted to investigate all adult individuals (DBH > 10 cm) growing in this area, however, in 2012 we could only locate 172 living trees, which were sampled for this study. All the sampled individuals were grouped into 5 spatially distinct populations (Tab. 1, Fig. 1). Rogozno and Lysakowo were the largest populations, consisting of 75 and 49 individuals, respectively. They were located along the edges of steep river valleys, mostly of southern exposure. The Zarosle and Osa Valley populations were distinctly smaller (7 and 16 individuals, respectively) and were located on a relatively flat area. The population Dusocin (25 trees) consisted of isolated individuals or small groups of trees (<5) growing usually on small forest hills ([8], [71]). Each sampled individual was georeferenced using GPS mapping system Pathfinder® ProXT™ (Trimble, Sunnyvale, USA) and the resulting data were visualized using the QGIS 2.8.2 free software (⇒ http://www.qgis.org/en/site/).
Tab. 1 - Extent of clonality in populations of Sorbus torminalis in the Forest District Jamy, Poland. (N): Number of ramets (trees); (G): number of genets; (N/G): mean number of ramets/genet; (Nc): number of clonal groups; (R): clonal richness [= (G-1)/(N-1)]; (1-R): extent of clonality.
| Population | N | G | N/G | Nc (no. ramets/group) |
R | (1-R) |
|---|---|---|---|---|---|---|
| 1. Dusocin | 25 | 19 | 1.316 | 4 (2, 2, 3, 3) |
0.750 | 0.250 |
| 2. Zarosle | 7 | 7 | 1.000 | 0 | 1.000 | 0.000 |
| 3. Rogozno | 75 | 30 | 2.500 | 12 (2, 2, 2, 2, 2, 3, 3, 4, 4, 4, 7, 17) |
0.392 | 0.608 |
| 4. Lysakowo | 49 | 31 | 1.581 | 8 (2, 2, 2, 3, 3, 4, 5, 5) |
0.625 | 0.377 |
| 5. Osa Valley | 16 | 13 | 1.231 | 1 (4) |
0.800 | 0.200 |
| All | 172 | 100 | 1.720 | 25 | 0.579 | 0.421 |
Fig. 1 - Distribution of populations of Sorbus torminalis at the north-eastern range limit. Populations are indicated by different colours and the numbers in parentheses indicate the population size (number of trees - Tab. 1).

Laboratory analyses
Total genomic DNA from 30 mg of dried leaf tissue was extracted according to the CTAB method ([18]), after grinding frozen tissue with Mixer Mill (MM301, Retsch, Haan, Germany). We screened all samples for variation at thirteen nuclear microsatellite markers: Sa01, Sa07, Sa14 ([29]); Mss1, Mss4, Mss5, Mss6, Mss9, Mss13, Mss16 ([55]); CH01h01 ([28]); CH02c09, Ms14H03 ([49]). Two PCR multiplexes were designed and optimized including six (multiplex-I: Ch01h01, Ch02c09, Mss4, Sa01, Sa07, Sa14) and seven loci (multiplex-II: Ms14H03, Mss1, Mss5, Mss6, Mss9, Mss13, Mss16). Details about the primers are presented in the Supplementary material (Tab. S1). The PCR mixture (10 μl in volume) consisted of 10 ng of template DNA, 1x of Multiplex PCR master mix (Qiagen), 0.5 mg/ml BSA, primers and water (Tab. S2 in Supplemetary material). Touch-down PCR was employed with cycling of 95 °C for 15 min followed by: 10 cycles of 94 °C for 30 s, 60 °C for 90 s with a reduction in temperatures of 0.5 °C per cycle, 72 °C for 60 s; then 25 cycles of 94 °C for 30 s, 55 °C for 90 s, 72 °C for 60 s; and a final cycle of 72 °C for 10 min. PCR reaction was conducted with PTC200® thermal cycler (MJ Research Inc., St. Bruno, Quebec, Canada). PCR products were sized using a capillary sequencer ABI PRISM 3130XL® (Applied Biosystems, Foster City, CA, USA). Allele calling was performed using GENESCAN 3.7 and GENOTYPER 3.7 software provided by Applied Biosystems. Allele binning was done manually after plotting of fragment size distributions for each locus ([32]).
Statistical analyses
Genotypes of individual trees were analyzed for the presence of replicates (ramets within a clone) using the Identity Analysis option of the CERVUS program v. 3.0.3 ([41]). Individuals with the same genotypes or with genotypes differing at one or two alleles were considered to belong to the same clone or genet (multi-locus lineage, MLL - [4]), and such one or two allele differences within MLLs were considered to be the results of somaclonal mutations. Clonal diversity (G/N), clonal richness [R = (G-1)/(N-1)] and the extent of clonality (1-R) were calculated according to Arnaud-Haond et al. ([4]), being G the number of genets and N the number of individual trees sampled. For subsequent population genetic analyses, only one genotype per clone was considered, regardless of the number of ramets within a clone. The consensus (most frequent) genotype within a genet was considered as representative for a given MLL.
Standard population genetic parameters were estimated for each locus, including: number of alleles (A), effective number of alleles (Ae), allelic richness (AR), observed (Ho) and expected (He) heterozygosity. The loci were also tested for the presence of null alleles, with their frequencies estimated jointly with FIS (Wright’s fixation index) using the IIM approach in INEST 2.0 software ([12]). Genetic differentiation between populations was assessed by estimating FST and RST with FSTAT v. 2.9.3.2 software ([30]).
We used two procedures to infer genetic structure of the studied populations. First, we performed the spatial mixture analysis with correlated allele frequencies implemented in BAPS v. 6 software ([13], [14]). We used an admixture model based on mixture clustering, and ran the analyses for 1000 iterations, setting the maximum possible number of subpopulations from 1 to 10. Second, we used a model-based clustering algorithm implemented in the STRUCTURE v. 2.3.4 software ([62], [24], [25]) to determine the number of subpopulations (K). We ran STRUCTURE using the default model parameters and varying K from 1 to 10. Each run consisted of 50.000 burn-in iterations and 100.000 data collection iterations, and was replicated 10 times. The number of clusters that best describes the data structure was found based on ΔK statistics ([23]) using the software package STRUCTURE HARVESTER ([19]).
Spatial genetic structure (SGS) within populations was investigated using the package SPAGeDi v. 1.4 ([33]). Due to small sample sizes within the populations, the SGS analysis was done for all populations jointly; however, only within populations pair-wise comparisons were allowed. The coordinates of a genet were determined as the arithmetic mean of coordinates of all individuals belonging to a given genet. The kinship coefficients (Fij - [50]) between pairs of individuals (or genets) were computed relative to local (within population) allele frequencies as suggested by Wang ([77]), and they were regressed on the logarithm of spatial distance to estimate the logarithmic regression slope blog. The significance of blog was tested based on 10.000 permutations of spatial positions of individuals within populations. The extent of SGS was estimated using the Sp statistic following Vekemans & Hardy ([75]). Sp was quantified by Sp = blog/[1 F(10,m)], where blog is the regression slope and F(10,m) is the mean kinship coefficient between individual pairs within the first distance class (0-10 m).
We used the ML-RELATE computer program ([40]) to infer sibling relationships among individual genotypes. This analysis was performed for the whole population jointly, and separately for each population and each cluster identified with BAPS software. We estimated the numbers of parent-offspring, full-sib and half-sib relationships in each group.
Results
Clonal structure
The 172 ramets (N) screened for variation at 13 nSSR loci were found to cluster into 118 unique multilocus genotypes. However, in a few cases individual genotypes differed by only one (15 cases) or two alleles (3 cases). Individuals with genotypes differing at one or two alleles were considered to belong to the same clone (multi-locus lineage, MLL - [4]), which suggested the existence of 100 clones (G) in the study area (G/N = 0.58 - see Tab. S3 in Supplementary material). Thus approximately 42% of trees belonged to clonal groups. For subsequent genetic analyses the consensus (most frequent) genotype was considered as a representative one for a given MLL (or genet).
The distribution of clonality was not uniform among populations (Tab. 1). There was no evidence of clonality in Zarosle, only one clonal group in Osa Valley (4 ramets) and just four clonal groups (2-3 ramets per clone) in Dusocin. In contrast, clonality was extensive in Rogozno and Lysakowo (12 and 8 clonal groups, respectively). In all cases, ramets of the same clone were growing in close proximity.
There was a strong correlation between the number of individual trees and the number of MLLs per population (r = 0.918, p = 0.028). The number of individual trees and the number of MLLs were significantly correlated (p < 0.05) to all clonal measures (number of clonal groups, clonal richness, extent of clonality). For example, the number of clonal groups within population was tightly correlated to the number of individual trees (r=0.994, p<0.001).
Notably, the largest clone was found in Rogozno and was composed by 17 individual trees (ramets), with DBH ranging from 7 to 17 cm. The distance between the most remote individuals within this clone was found to be 21 meters. Interestingly, the tree considered as the largest wild service tree in Poland (DBH = 70 cm) growing in Dusocin ([8]) appeared to be just one of the three ramets of a clone, indicating that the individual as a genet is even larger in size than previously considered.
Somaclonal variation
Comparing genotypes within each MLL, we found 20 cases of allelic differences observed within 16 MLLs. These differences were considered as the effect of somaclonal mutation. When mutated genotypes were compared with consensus genotypes within MLL, the mutation usually affected a larger allele of a pair of alleles within a locus of a given individual (16 cases), with only 4 cases of a smaller allele being mutated. The mutations resulted usually in an increase of the fragment size, with the changed allele being on average greater by 2 bp (one repeat unit), as compared to the consensus one. Somaclonal variation was noted in 6 out of 13 loci. The largest number of mutations (10) was observed in locus Mss16. The overall frequency of mutations measured within MLLs was 0.0084 (or 0.0045 when excluding the locus Mss16), with the most variable locus Mss16 having the mutation frequency of 0.0544.
Genetic diversity
Genetic diversity parameters were calculated based on the consensus genotypes established for each clone. Thus, the analyses of genetic diversity and populations structure were calculated based on a set of 100 distinct genotypes. All 13 microsatellite loci appeared to be polymorphic with the number of alleles at each locus ranging from 4 to 19, with a mean of 10 (Tab. 2). Average effective number of alleles was equal to 4.939. Average observed and expected heterozygosities were found to be 0.777 and 0.756, respectively. Wright’s fixation index (Fis) was found to be -0.029, indicating a small excess of heterozygotes. A detectable proportion of null alleles was found at loci Mss4, Sa01, Mss1 and Mss9, but in all cases they were not significantly different from 0. No deviations from Hardy-Weinberg equilibrium were found at any locus.
Tab. 2 - Genetic diversity of the whole population of Sorbus torminalis in the Forest District Jamy, Poland. (A): mean number of alleles; (Ae): effective number of alleles; (AR): allelic richness; (Ho): observed heterozygosity; (He): expected heterozygosity; (Fis): inbreeding coefficient; (Null): frequency of null alleles; (H-W): significance of departure from Hardy-Weinberg equilibrium; (ns): not significant (p>0.05).
| Locus | A | A e | AR | H o | H e | F is | Null | H-W |
|---|---|---|---|---|---|---|---|---|
| Ch01h01 | 10 | 6.974 | 9.980 | 0.850 | 0.855 | 0.006 | 0 | ns |
| Ch02c09 | 8 | 4.296 | 8.000 | 0.900 | 0.766 | -0.176 | 0 | ns |
| Mss4 | 4 | 2.218 | 4.000 | 0.610 | 0.625 | 0.024 | 0.014 | ns |
| Sa01 | 12 | 5.663 | 11.979 | 0.730 | 0.805 | 0.093 | 0.023 | ns |
| Sa07 | 5 | 2.483 | 5.000 | 0.667 | 0.605 | -0.101 | 0 | ns |
| Sa14 | 18 | 4.000 | 17.969 | 0.820 | 0.764 | -0.074 | 0 | ns |
| Ms14H03 | 7 | 3.804 | 6.990 | 0.730 | 0.724 | -0.008 | 0 | ns |
| Mss1 | 9 | 3.865 | 7.980 | 0.760 | 0.738 | -0.030 | 0.013 | ns |
| Mss13 | 7 | 2.473 | 6.980 | 0.620 | 0.600 | -0.034 | 0 | ns |
| Mss16 | 19 | 11.614 | 18.979 | 0.930 | 0.923 | -0.008 | 0 | ns |
| Mss5 | 10 | 5.701 | 9.990 | 0.830 | 0.817 | -0.016 | 0 | ns |
| Mss6 | 10 | 4.829 | 9.980 | 0.830 | 0.784 | -0.059 | 0 | ns |
| Mss9 | 17 | 6.588 | 15.980 | 0.830 | 0.817 | -0.016 | 0.006 | ns |
| Mean | 10 | 4.939 | 10.293 | 0.777 | 0.756 | -0.029 | 0.004 | - |
| SE | 1.338 | 0.762 | 1.315 | 0.028 | 0.027 | 0.018 | 0.002 | - |
Genetic structure (clustering)
The differentiation among populations was rather low with average FST = 0.0477 ± 0.0058 (SE) and RST = 0.0588 ± 0.0100. We found no relationship between genetic distance among pairs of populations measured as FST/(1-FST) and their physical distance quantified as logarithm of distance. However, both BAPS and STRUCTURE clustering methods identified three distinct clusters as the most likely structure of the analyzed data set. Notably, there was a high congruence between the two methods, since 90% of genets were assigned to the same clusters in each of the clustering methods (Fig. 2). Therefore, we present below only the result of clustering identified with BAPS software.
Fig. 2 - Spatial distribution of clusters identified using the BAPS software (cluster 1: red; cluster 2: green; cluster 3: blue). The size of the circles is proportional to the number of clones within population (Tab. 1).
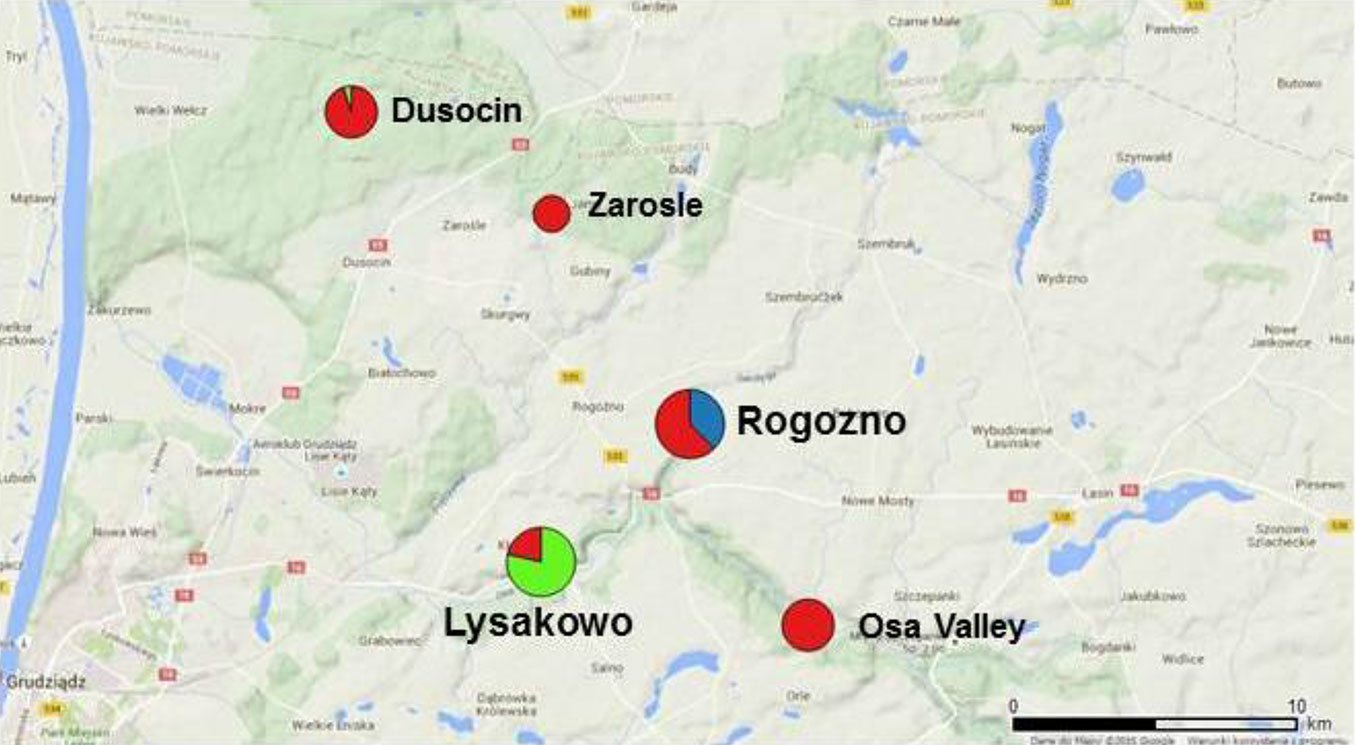
The smallest cluster (cluster 3 - Tab. 3) was composed of 9 genets only. It appeared to be spatially restricted and fully included in the population Rogozno. Cluster 2 consisted of 22 individuals from population Lysakowo and one isolated remote individual assigned to population Dusocin. All the remaining 68 genets were grouped into the largest cluster 1. Although, the cluster 1 was spread across all populations, the cluster 2 and 3 were spatially restricted.
Tab. 3 - Probabilities of relationships for different populations and clusters identified with BAPS software, estimated based on pairs of individuals.
| Group | Pop | Sample size |
Full sibs (%) |
Half sibs (%) |
Full + Half sibs (%) |
Parent/ Offspring (%) |
Unrelated (%) |
|---|---|---|---|---|---|---|---|
| Population | Dusocin | 19 | 1.2 | 9.3 | 10.5 | 0 | 89.5 |
| Zarosle | 7 | 0 | 0 | 0 | 0 | 100.0 | |
| Rogozno | 30 | 1.8 | 6.2 | 8.0 | 3.7 | 88.3 | |
| Lysakowo | 31 | 3.4 | 10.8 | 14.2 | 4.1 | 81.7 | |
| Osa Valley | 13 | 2.6 | 5.1 | 7.7 | 3.8 | 88.5 | |
| Mean | 20.0 | 1.8 | 6.3 | 8.1 | 2.3 | 89.6 | |
| BAPS cluster | 1 | 68 | 0.6 | 11.1 | 11.7 | 0.6 | 87.7 |
| 2 | 23 | 5.1 | 9.9 | 15.0 | 4.3 | 80.7 | |
| 3 | 9 | 2.8 | 22.2 | 25.0 | 11.1 | 63.9 | |
| Mean | 33.3 | 2.8 | 14.4 | 17.2 | 5.3 | 77.5 | |
| All | Total | 100 | 1.3 | 11.7 | 13.0 | 0.9 | 86.1 |
Spatial genetic structure within populations
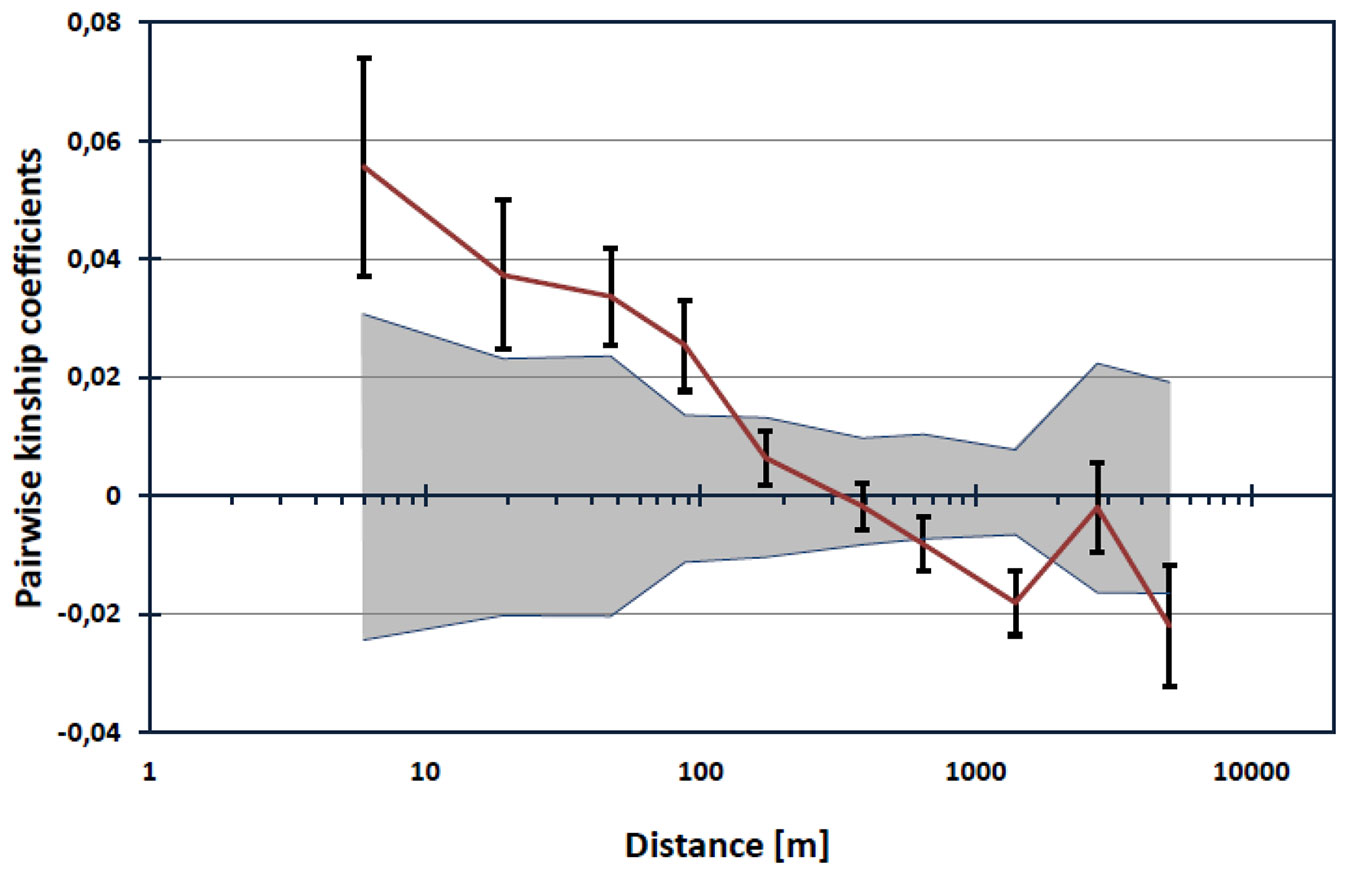
We found evidence of spatial genetic structure within populations. Pairwise kinship coefficients appeared to be significant for pairs of individuals separated up to about 100 meters (Fig. 3). The slope of the regression of the kinship coefficient on the logarithm of spatial distance between genets was found to be blog = -0.012 (p<0.001). The Sp statistic was 0.0125, indicating a weak but significant fine-scale pattern of isolation by distance.
Fig. 3 - Autocorrelogram of estimated kinship coefficients (Fij) against the logarithm of distance. Upper and lower 95% confidence intervals around the null distribution of kinship coefficients derived from 10.000 permutations are indicated by shaded area. Black bars around Fij values represent standard errors obtained by a jackknife procedure over loci.
Relatedness
The levels of relatedness varied among populations. There was no relatedness among individuals in the population of Zarosle, which is composed of scattered individuals only. The highest level of relatedness in all categories was found in Lysakowo (Tab. 3). Other populations exhibited intermediate and comparable levels of relatedness (Tab. 3), however, we found no indication of parent / offspring relationships in Dusocin. When considering clusters of individuals identified with BAPS software, the highest level of relatedness was found in the cluster 3, where only 64% pair-wise comparisons indicated lack of relatedness. However, this cluster is relatively small (9 MLLs) and spatially restricted, and belongs to the population Rogozno (Fig. 3). The largest cluster (No. 1) composed of 68 MLLs, embracing individuals from every population, exhibited the lowest level of relatedness (Tab. 3). The remaining cluster 2, being the spatially restricted part of the population Lysakowo (Fig. 3), had the largest proportion of fullsib relationships, and the proportion of pairwise comparisons - indicating lack of relatedness - was found to be 80.7%. It should be noted that we found generally positive correlations between the number of MLLs and relatedness measures, but they appeared to be insignificant, likely due to a small number (5) of compared populations (e.g., proportion of fullsibs + halfsibs: r=0.788, p=0.114).
Discussion
Mapping genetic diversity and structure in marginal plant populations is essential for managing their genetic resources. This is particularly important for rare, scattered tree species, such as Sorbus torminalis. For example, such knowledge allows to design seed sampling for ex-situ and in-situ conservation efforts (selection of populations and spacing among sampled trees) maximizing genetic diversity of future generations. Because S. torminalis has the ability for vegetative reproduction and clonal growth ([35], [66]), population genetic surveys of this species should be conducted with careful assessment of the presence of clones, which should be taken it into account in subsequent genetic analyses ([4]). This is particularly important in Sorbus species and other members of the Rosaceae family, which are known for their capacity for clonal propagation through rootsuckers ([53], [64], [35], [65], [74], [38]).
Clonality in plants is often considered as an effective strategy for survival and the maintenance of existing genetic diversity of populations during harsh environmental conditions or during colonization of new areas where sexual reproduction is limited ([73]). This strategy seems to be particularly useful in plants with self-incompatibility systems. Vegetative propagation may continue until mating among unrelated individuals becomes possible, producing new generations of variable offspring cohorts ([73]).
We found the mean number of ramets per genet (1.72 - Tab. 1) quite comparable to that reported for other populations across the northern range of this species ([57], [35], [66]). The distribution of clonality among populations investigated in this study, however, was not uniform. Clones were more frequent in populations growing on rugged areas, such as steep river valleys or mid-forest hills. Fairly extensive clonality was noted in peripheral northern populations growing at the Baltic coast of Denmark and Germany, with on average 2.64 ramets per genet ([66]). On the other hand, 2.26 ramets per genotype were found in Switzerland, but there were populations with some clones represented by more than 10 ramets ([35]). Other authors reported only scarce examples of clonality. For instance, only few stems over 185 individuals were found to be the result of vegetative reproduction in a French population of S. torminalis ([57]). Some studies may underreport the natural incidence of clonality because they were specifically designed to avoid sampling from the same genets, which prevented precise estimation of the extent of clonality ([11]).
Somatic mutations are increasingly recognized as an inevitable feature in clonally reproducing plants and long-lived tree species ([52], [31], [37]). Somatic mutations result in minor DNA variation within clonal lineages ([37]) and it is suggested that old clones may accumulate somatic mutations to a greater extent ([79], [72], [52]). Understanding the development of somaclonal variation requires closer attention because older and slowly-growing clones could have levels of mutations similar to those of younger, faster-growing clones, as the mutation rate is expected to be proportional to the number of mitotic divisions ([43], [1], [2], [37]). As somatic mutations accumulate in clones, the remaining stems may appear to be genetically distinct clones. Somatic mutation at microsatellite loci have been observed in the Rosaceae family: Prunus ssiori ([53]), P. avium ([74]), but also in other plants: Fragaria virginiana ([78]), Grevillea renwickiana, ([37]), G. rhizomatosa ([31]), including tree species: Robinia pseudoacaccia ([48]), Populus euphratica ([68]), Populus tremuloides ([52]), Quercus robur ([6]), Quercus ilex ([59]), Thuja plicata ([61]). Somaclonal variation of microsatellite loci may confound the inference of clonal structure ([4]), but usually a careful assessment of allelic variation among putative ramets of the same clone enable the proper assignment of specimens to respective clones, thus establishing multi-locus lineages (MLLs - [4], [74]). The somaclonal variation detected in this paper (i.e., most frequent allele change of one 2-bp microsatellite repeat unit) seem to be in line with the expected microsatellite mutation models ([22]).
Although we found extensive clonality of S. torminalis, we also found relatively high levels of genetic diversity across the surveyed region. This contrasts the results of a study on S. torminalis populations growing at the northern range limit by Rasmussen & Kollmann ([66]), who found extensive clonality and low genetic diversity. The authors also observed high genetic differentiation among populations (FST = 0.341), which was partly attributed to extensive clonality and genetic drift. Our results showed similar numbers of alleles per locus as reported in more central populations of France ([55]) and Switzerland ([42]) and higher genetic diversity than that observed in S. domestica across Europe ([27]).
Inbreeding coefficients of S. torminalis are usually found to be slightly negative, indicating excess of heterozygotes as compared to expectations from Hardy-Weinberg equilibrium ([10], [36]). Our results confirmed this finding with inbreeding coefficients being consistently slightly negative across almost all SSR loci. This is reasonable given the self-incompatibility mating system of the species ([51], [70]); however, asexual reproduction may also affect FIS estimates ([69]).
Although genetic diversity is expected to decrease towards species distribution margins ([20]), there are several examples where genetic diversity within populations did not decline towards range margins, but an increase of population differentiation was observed ([80], [76], [5]). This may be particularly relevant for S. torminalis. Kučerová et al. ([46]) found higher differentiation over central-European populations than those located in southern locations. Genetic differentiation among populations investigated in this study was found to be low, as compared to other studies ([15], [10], [3], [46]), which might be attributed to close proximity of the populations under study. The FST and RST estimates were quite similar (FST = 0.048 ± 0.006 and RST = 0.059 ± 0.010), indicating that the differentiation was not caused by mutation, but rather it was due to specific colonization and gene flow patterns among populations ([7]).
With the climate changes predicted in the future, S. torminalis is expected to extend its range towards northern and eastern locations ([60]). The studied populations with their high genetic diversity may thus become an important source of genetic diversity for natural or assisted expansion of the species into new areas where climate conditions will become more favorable ([45]).
Genetic structure among populations based on Bayesian clustering methods revealed only moderate population structuring, which seems to be in line with the assessment of among population differentiation. However, we identified significant spatial genetic structure within populations, indicating that at a local scale, isolation by distance may be an important mechanism shaping the distribution of genetic diversity (Fig. 3). The spatial genetic structure parameter Sp ([75]), was found to be Sp = 0.0125, and it appeared similar to that reported in a French population (Sp = 0.0187) by Oddou-Muratorio & Klein ([58]). However, spatial genetic structure was not found in Swiss populations ([35]). Our results are generally in line with the expectations for clonal insect pollinated and animal seed-dispersed species, as recently reviewed by Dering et al. ([17]).
Genetic diversity in peripheral populations may be determined by locally specific factors such as the history of colonization and local population dynamics (e.g., expansion or shrinkage). The spatial distribution of genetic variation observed in this study seems to be the result of specific pattern of colonization of the studied area, as revealed by the distribution of relatedness among populations or groups identified by clustering methods (Tab. 3). However, the subsequent populations expansion could have led to the existing within-population spatial genetic structure. The relatively high genetic diversity of the populations seems to be maintained by efficient gene dispersal coupled with a self-incompatibility mating system ([35], [3],). Indirect estimates of relative pollen to seed gene dispersal in S. torminalis have been found to be low (1.84-2.92 - [3], [58]), indicating the importance of seed dispersal in shaping the genetic diversity in this species. Efficient pollen and seed dispersal over distances greater than 10 km have been detected in S. domestica, a species with similar pollination and seed dispersal biology ([42]), suggesting that fragmented populations, such as those investigated in this study, may nonetheless be genetically connected by gene flow.
Our study demonstrated that peripheral populations of S. torminalis may have relatively high levels of genetic diversity, despite the occurrence of clonal propagation. The studied populations demonstrated low genetic differentiation and lack of inbreeding. However, the existence of clonality and signatures of spatial genetic structure should be taken into consideration when planning seed harvesting of this protected species for future conservation genetic programs in the studied area and in other similar populations. Avoiding sampling seeds from nearby trees, likely related or being even ramets of the same clone, should promote high genetic diversity of resulting progeny generations.
The frequency of clonal versus sexual reproduction across the range of S. torminalis is not fully characterized, but population surveys done to date suggest that the predominant mode of reproduction within a population changes across the species range. The propensity for clonal reproduction is likely to be determined by local (e.g., topography) conditions, although the factors influencing the tendency for clonal versus sexual reproduction are unknown. Because the extent of clonal reproduction varies between populations of S. torminalis, it is very likely that the spatial genetic structure within and among populations will also vary. An effective management strategy for this species should incorporate the variability in reproductive biology of this species and thus requires a broader survey of the genetic variation spanning over the species’ range.
Acknowledgements
We thank Dr. Magdalena Bartkowska for valuable comments and suggestions, which significantly improved the paper. The study was supported by the National Science Center, Poland (Grant No: 2011/03/ B/NZ9/03139). Plant material was sampled based on the permission WPN.6402.1.13. 2011.WR issued by the Regional Directorate for Environmental Protection in Bydgoszcz, Poland. The authors are thankful to Igor Chybicki for his help in collecting samples in the field. Local support and field guidance by Andrzej Tarnawski and Tadeusz Kempa from the Directorate of Forest District Jamy is greatly acknowledged.
SJ-W and JB planned the research, contributed to statistical analyses and wrote the article; SJ-W, KM and ES designed and optimized PCR multiplexes and performed laboratory analyses; LK and JB designed the sample collection and sampled the plant material; JB obtained funding.
References
Gscholar
Gscholar
CrossRef | Gscholar
CrossRef | Gscholar
Gscholar
Gscholar
CrossRef | Gscholar
Authors’ Info
Authors’ Affiliation
Katarzyna Meyza
Ewa Sztupecka
Lukasz Kubera
Jaroslaw Burczyk
Institute of Experimental Biology, Department of Genetics, Kazimierz Wielki University of Bydgoszcz, Chodkiewicza 30, 85-064 Bydgoszcz (Poland)
Corresponding author
Paper Info
Citation
Jankowska-Wroblewska S, Meyza K, Sztupecka E, Kubera L, Burczyk J (2016). Clonal structure and high genetic diversity at peripheral populations of Sorbus torminalis (L.) Crantz.. iForest 9: 892-900. - doi: 10.3832/ifor1885-009
Academic Editor
Andrea Piotti
Paper history
Received: Sep 27, 2015
Accepted: Feb 02, 2016
First online: May 29, 2016
Publication Date: Dec 14, 2016
Publication Time: 3.90 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2016
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 54620
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 44669
Abstract Page Views: 4298
PDF Downloads: 4153
Citation/Reference Downloads: 68
XML Downloads: 1432
Web Metrics
Days since publication: 3724
Overall contacts: 54620
Avg. contacts per week: 102.67
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Jul 2026)
Total number of cites (since 2016): 17
Average cites per year: 1.55
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Analysis of growth of recruits of natural regeneration of Sorbus torminalis (L.) Crantz - a rare European forest tree species
vol. 11, pp. 72-78 (online: 25 January 2018)
Research Articles
Chloroplast microsatellites as a tool for phylogeographic studies: the case of white oaks in Poland
vol. 8, pp. 765-771 (online: 19 July 2015)
Research Articles
Arbuscular mycorrhizal fungal symbiosis with Sorbus torminalis does not vary with soil nutrients and enzyme activities across different sites
vol. 8, pp. 308-313 (online: 03 September 2014)
Research Articles
Comparison of range-wide chloroplast microsatellite and needle trait variation patterns in Pinus mugo Turra (dwarf mountain pine)
vol. 10, pp. 250-258 (online: 11 February 2017)
Research Articles
Seedling emergence capacity and morphological traits are under strong genetic control in the resin tree Pinus oocarpa
vol. 17, pp. 245-251 (online: 16 August 2024)
Research Articles
Genetic variation and heritability estimates of Ulmus minor and Ulmus pumila hybrids for budburst, growth and tolerance to Ophiostoma novo-ulmi
vol. 8, pp. 422-430 (online: 15 December 2014)
Commentaries & Perspectives
The genetic consequences of habitat fragmentation: the case of forests
vol. 2, pp. 75-76 (online: 10 June 2009)
Research Articles
Patterns of genetic variation in bud flushing of Abies alba populations
vol. 11, pp. 284-290 (online: 13 April 2018)
Research Articles
Genetic diversity of core vs. peripheral Norway spruce native populations at a local scale in Slovenia
vol. 11, pp. 104-110 (online: 31 January 2018)
Review Papers
Genetic diversity and forest reproductive material - from seed source selection to planting
vol. 9, pp. 801-812 (online: 13 June 2016)
iForest Database Search
Search By Author
Search By Keyword
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords
PubMed Search
Search By Author
Search By Keyword







