Adaptive response of Pinus monticola driven by positive selection upon resistance gene analogs (RGAs) of the TIR-NBS-LRR subfamily
iForest - Biogeosciences and Forestry, Volume 10, Issue 1, Pages 237-241 (2017)
doi: https://doi.org/10.3832/ifor2050-009
Published: Feb 02, 2017 - Copyright © 2017 SISEF
Research Articles
Abstract
Western white pine (Pinus monticola Dougl.) is an important forest tree species, which is intensively plagued by the fungus Cronartium ribicola. Resistance gene analogs (RGAs) are the most highly abundant class of potential resistance (R) genes sharing greatly conserved domains and structures. Hence RGAs are crucial components for disease resistance breeding programs on P. monticola serving as useful functional markers. A total of 33 P. monticola RGAs gene homologues were mined from GenBank, encoding for R gene members of the TIR-NBS-LRR subfamily. The existence of positive selection acting upon RGAs was determined using a series of maximum likelihood analyses. Robust evidence of positive selection was showed to be acting widely in three clades across RGA gene phylogeny, both on terminal and ancestral lineages. Furthermore, our analysis revealed that the majority of positively selected residues sites are localized widely across these RGAs sequences, putatively affecting the structures of their ligand-binding domains and offering novel specificities. These results may find immediate application in ongoing disease resistance breeding programs.
Keywords
Fungal Diseases, Genomics-assisted Breeding, Non-synonymous Nucleotide Substitution, Positive Selection, Resistance Gene Analogs (RGAs), White Pines
Introduction
Western white pine (Pinus monticola Dougl.) is an ecologically dominant, and highly important for forestry, species in the western North American ecosystems ([20]). In 1980, two P. monticola provenances from Jackson, Oregon were introduced in Greece in comparative species field trials of the Aristole University of Thessaloniki, Laboratory of Forest Genetics Experimental Centers located in Pertouli and Taxiarchis ([26], [27]). The aim of this P. monticola introduction was twofold: (a) the use of the species in plantation forestry for wood production, and (b) the evaluation of resistance to the white pine blister rust (Cronartium ribicola J.C. Fisch.). Trial results indicate large variation within provenance, growth comparable to that of local Abies borisii regis Mattf., but reduced survival ([28], [27]).
Cronartium ribicola is among the most destructive white pine fungal diseases which has caused severe mortality in North American white pines and especially in P. monticola, which is currently considered a species under threat ([21]). P. monticola species exhibits genetic variation in disease resistance to C. ribicola involving mainly a dominant disease resistance gene (Cr2 - [12], [19]). Disease resistance and maintenance of genetic diversity in production populations are very important aspects for P. monticola breeding ([4]). The development through breeding of trees resistant to fungi and the maintenance of a variety of genetic resources are of pivotal significance towards the substantial establishment of P. monticola reforestation and plantation forestry programs ([18]).
Classical breeding approaches, such as interspecific hybridization with the indigenous to Greece and resistant to C. ribicola Balkan white pine Pinus peuce Grisb., are time-consuming without warranties of specific results in a definite timeframe. Engagement of innovative molecular breeding approaches allows breeders to establish sustainable resistance more accurately and at a faster pace ([14]). PCR approaches implementing mainly degenerate primers were extensively applied in order to isolate members of the NBS family (nucleotide-binding site) / LRRs (Leucine-Rich Repeats) - containing genes, the most highly expanded group of genes linked directly to resistance (R) genes mediated functions in plants ([5]). This protein family is further classified into two major subfamilies based on structural characteristics of their putative amino-terminal signaling domain ([13]). One subfamily, TIR-NBS-LRR, contains an amino-terminal signaling domain that depicts homology with Toll and interleukin- 1 receptor (TIR), whilst the other subfamily, CC-NBS-LRR, instead of the TIR domain, carries a putative coiled-coil structure (CC) one ([13]). Both these subfamilies have been reported to mediate resistance to a wide range of phytopathenic fungi ([34]).
Resistance gene analogs (RGAs) are a large class of potential R-genes, being an important resource for the development of molecular markers, hence enhancing numerous disease resistance breeding efforts ([1], [30]). Moreover, these RGAs commonly contain tandem repeats such as LRRs which give rise to novel ligand-binding specificities under positive selection ([11]) via a number of different modes, such as point mutations, variations in repeat numbers and gene duplications ([8], [24], [37], [39]). Positive selection is defined as the rapid fixation of beneficial non-synonymous mutations and is an important evolutionary driving force for a number of paralogous genes towards their functional diversification, which might be selectively favored under biotic pressures in plants ([6]). Furthermore, the location of positively selected amino acid residues is an important aspect for obtaining novel gene functions ([25]). Previous studies have reported that solvent-exposed regions of the LRRs repeats are subjected to positive selection; such evidence has been interpreted as a sign of the involvement of these regions in pathogen recognition ([29], [39], [11]). Overall, the evolution patterns of plant NBS-LRR R genes and of their RGAs counterparts is likely a complex process ([40], [31]).
The TIR-NBS-LRR RGAs from P. monticola were previously investigated for their diversity and expression, in order to gain insights regarding their potential usage in disease resistance breeding programs ([17]). In this study, we address the question whether signatures of positive selection are systematically employed as an evolutionary driven force acting on TIR-NBS-LRR-encoding RGAs genes at P. monticola. Thus, we gain insights into the evolutionary profiles of the P. monticola TIR-NBS-LRR-containing RGAs genes, hypothesizing that those successive episodes of positive selection might contribute in the acquisition of novel pathogens recognition repertoires. Overall, our findings enrich and support the hypothesis that these P. monticola RGAs could be employed as biomarkers for both, monitoring the ongoing and, more effectively applying future P. monticola disease resistance breeding efforts.
Materials and methods
P. monticola TIR-NBS-LRR RGAs mining and phylogenetic analysis
All 33 P. monticola RGAs partial proteins previously identified by Liu & Ekramoddoullah ([17]) were downloaded from the GenBank by keyword searches based on their NB-ARC domain profile (PF00931.19/ CL0023). Using these amino acid sequence IDs, the respective ORF nucleotide sequences were also retrieved. All amino acid sequences had to fulfill the following criteria in older to be evaluated for evolutionary signatures acting upon them: (a) existence of valid structures by means of protein lengths up to 90 residues, positions of NB-ARC domain motifs, blast(p) hits against known plant resistance genes; and (b) verification of functional domains using the predictions against the Pfam database (⇒ http://pfam.sanger.ac.uk/search#tabview= tab1) and the InterProScan 5 software ([9]).
The phylogenetic relationships among these P. monticola RGAs amino acid sequences were revealed by aligning them using the MUSCLE software ([7]) and performing a tree reconstruction using the RAxML software ([32]) inferring a gamma model of rate heterogeneity. All these analyses were performed on the Geneious R7 platform ([10]).
P. monticola TIR-NBS-LRR RGAs evolutionary analysis
The YN00, CODEML and CODEMLSITES programs were employed from the PAML version 4.8 software package ([36]) in order to assess evidence of positive selection signatures acting in these P. monticola TIR-NBS-LRR RGAs. These tests compare the substitution rates of non-synonymous (dN) to synonymous (dS) mutations, and estimate the derived omega (ω) values, both among the nucleotide codon sequences and along the branch lineages. The rates of dN/dS nucleotide substitutions per site across all possible pairwise sequence comparisons were estimated using the counting approach ([35]) implemented in the YN00 program. The total dN, dS, and ω values for all pairwise sequence comparisons among these RGAs are shown in Tab. S1 (Supplementary material).
An NJ tree at amino acid level and the respective nucleotide alignment were both used in the CODEML and CODEMLSITES analyses according to Lynn et al. ([22], [23]); details of the exact methods for these analyses are found in Zambounis et al. ([39]). Briefly, the amino acid sequence alignment was conducted by employing the MUSCLE software ([7]), whilst the construction of the respective phylogenetic tree was performed using the MEGA 5 software ([33]). A Poisson correction was employed with a gamma distribution modeling among sites and rates variations ([39]). Finally, the Baseml software (PAML version 4.8 - [36]) was implemented for estimating the alpha shape parameter using the REV model.
CODEML was tested in P. monticola TIR-NBS-LRR RGAs dataset for estimating the variable selective pressures acting among lineages in the phylogeny, whilst CODEMLSITES was employed to test for site-specific codon substitution models allowing the identification of selective signatures across amino acid residues (Tab. 1). In both programs log-likelihood calculations were performed for each model and comparisons between each other were implemented by likelihood ratio tests (LRTs). The empirical Bayesian method further allowed the detection of codon residues subjected to positive selection by higher posterior probabilities. All data sets as well as the outputs of CODEML and CODEMLSITES programs are available upon request.
Tab. 1 - Overview of CODEMLSITES method statistics and distribution of positively selected amino acid sites across TIR-NBS-LRR RGAs in P. monticola.
| Model | P | Parameters |
l
|
dn/ds | Positively Selected Sites |
Statistical significance |
|---|---|---|---|---|---|---|
| M0: one ratio | 2 | ω = 0.395 | -2855.76 | = ω | - | - |
| M1: neutral | 3 | p0 = 0.72, ω0 = 0.22 | -2821.47 | 0.44 | - | - |
| p1 = 0.27, ω1 = 1 | ||||||
| M2: selection | 5 | p0 = 0.72, ω0 = 0.22 | -2821.47 | 0.44 | - | P < 0.001 |
| p1 = 0.177, ω1 = 1 | ||||||
| p2 = 0.102, ω2 = 1 | ||||||
| M3: discrete | 6 | p0 = 0.201 ω0 = 0.048 | -2811.41 | 0.41 | 19, 50, 56, 69, 74, 79, 83 | P < 0.001 |
| p1 = 0.594, ω1 = 0.335 | ||||||
| p2 = 0.197, ω2 = 1.019 | ||||||
| M7: beta | 3 | p = 0.825, q = 1.26 | -2812.75 | 0.39 | - | - |
| M8: beta and ω | 5 | p0 = 0.923, p = 1.164, q = 2.28 |
-2817.16 | 0.41 | 19, 56, 69, 74, 79, 83 | P < 0.05 |
| p1 = 0.076, ω = 1.322 |
Results
P. monticola TIR-NBS-LRR RGAs structure and phylogenetic analysis
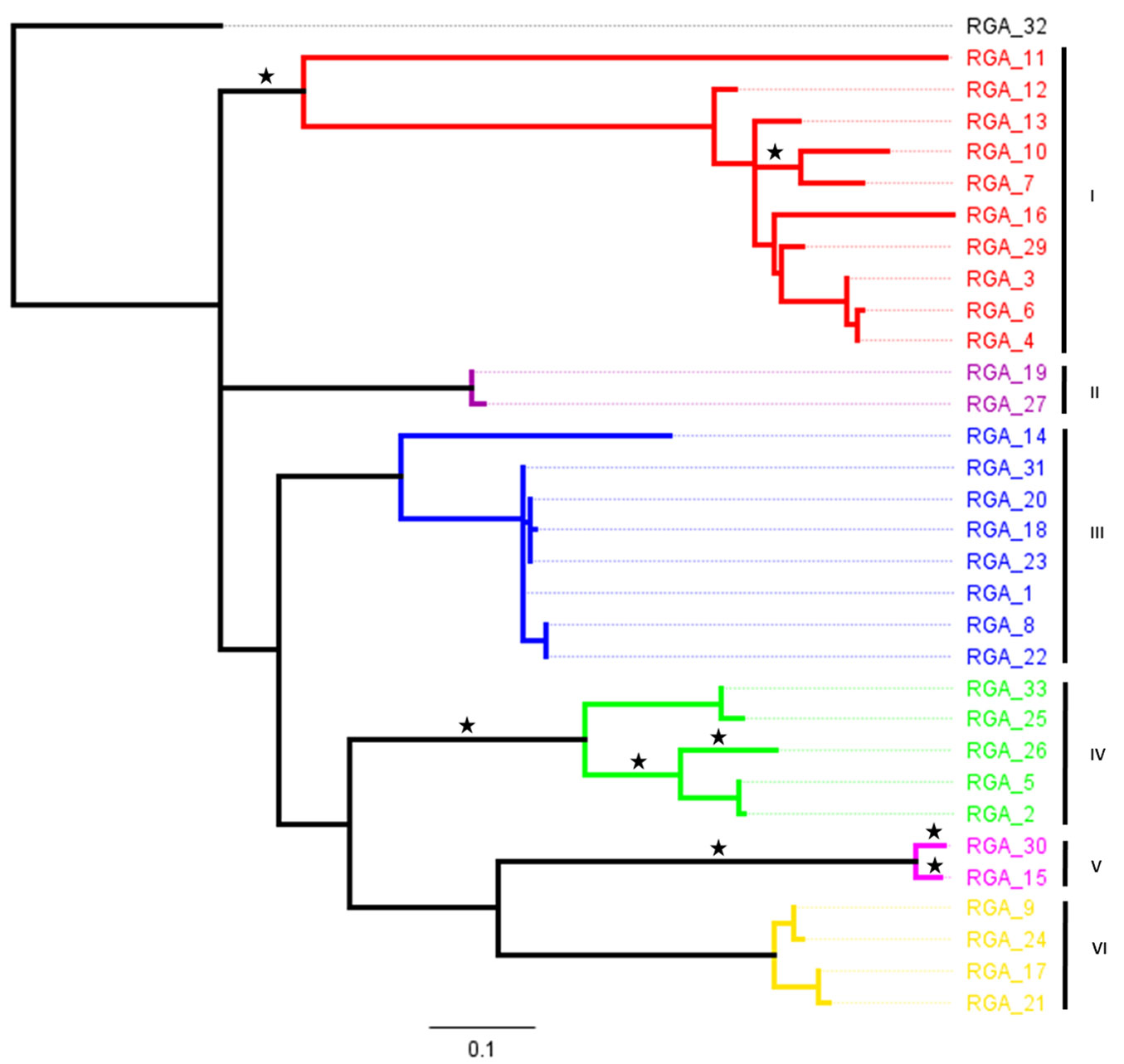
The 33 P. monticola TIR-NBS-LRR RGAs studied were found to share Pfam hits with the NB-ARC domain (PF00931.19) and the InterProScan ones with LRR-containing proteins (PTHR23155). The RGAs were corresponding to regions between conserved kinase-2 and P-loop domains. A RAxML phylogenetical analysis at the amino acid level was conducted in order to explore their expansion (Fig. 1). The backbone topology of this tree was properly resolved with 63 branches (Fig. 1), whilst the overall pairwise identity among the amino acid sequences was 59.6%, indicative of a high degree of divergence.
Fig. 1 - RAxML phylogenetic reconstruction of the amino acid sequences of P. monticola TIR-NBS-LRR RGAs. In various colors the six clades which were being assigned whilst, asterisks (*) indicate tree branches under positive selection (ω values higher than 1) with posterior probabilities using the CODEML method.
Positive selection acting across P. monticola TIR-NBS-LRR RGAs lineages
In order to verify whether accelerated evolution and signs of positive selection might have also contributed to the divergence of these TIR-NBS-LRR RGAs genes, our RGAs dataset was subjected to several successive tests of positive selection using the counting (YN00) and the ML (CODEML/ CODEMLSITES) methods. A robust and reliable nucleotide alignment resulted, based on the presence of zero or few gaps across the aligned sequences. The dN and dS mutation values across the entire RGAs sequences were calculated using the counting YN00 method (see Tab. S1 in Supplementary material). Any substitution saturation effects were checked by calculating dS rates between the aligned nucleotide sequences. The average numbers of synonymous (dS) mutations per synonymous sites over all sequence pairs were below the value of 2, the crucial threshold value above which the corresponding sequences would have to be excluded from further evolutionary analyses in order to bypass any saturation effects based on nucleotide substitutions ([35]).
Finally, the CODEML approach was employed in order to identify branches subjected to positive selection. Statistically significant evidence of positive selection (ω > 1.00) was detected in numerous branches (Fig. 1) with posterior probabilities P < 0.0001. These signs were found across clades I, IV and V acting with a non-general rule, as positive episodes were evidenced both in the ancient and terminal branches (reflecting probably a series of recently successive bursts of gene duplications).
Positive selective pressures acting among amino acid residues across the P. monticola TIR-NBS-LRR RGAs
Possible positive selection at the individual amino acid residues level was investigated by the CODEMLSITES method ([36]) which compared various models (M0 against M3, M1 against M2, and finally M7 against the stringent M8) in our RGAs dataset. Two models (M3 and M8) revealed positive selective pressures acting among amino acid residues under significant posterior probabilities (P < 0.0001 and P < 0.05, respectively - Tab. 1). These extensive signs of positive selection were found acting rather widely upon amino acid residues and across the respective tree branches. The accurate locations of these positively selected sites (with ω values up to 1.48) are depicted in Tab. 1.
Discussion
Conifer genomes are in general large with DNA content reaching more than 26.500 Mbp per haploid genome ([2] and references therein). Taking also into consideration their long generation times, classical breeding, especially for disease resistance is a very slow process. On the contrary, molecular breeding approaches may provide promising and effective strategies for both R genes and RGAs counterpart identification based on their highly conserved motifs ([20]). Previously, it was postulated that there is a highly diverse group of TIR-NBS-LRR genes in P. monticola ([17]) along with the existence of numerous CC-NBS-LRR-containing proteins ([20]). Similar abundance and diversity of these genes was also observed in other plant genomes ([40], [3], [31]). Hitherto, TIR-NBS-LRR RGAs in P. monticola exhibited high variation at amino acid levels, whilst almost half of them were found to be pseudogenes representing a rather complex TIR-NBS-LRR subfamily ([17]).
In our study, the analysis of 33 P. monticola TIR-NBS-LRR RGAs showed a high level of diversity that may contribute to their evolutionary fitness, and the putatively functional character of their transcription activity ([17]). These RGAs are a potential source for the development of molecular markers, especially regarding selection of resistance to white pine blister rust. Furthermore, RGAs were found to share conserved structures rendering them ideal candidates for applying evolutionary bioinformatics approaches.
The CODEML analysis, showed recent episodes of positive selection acting extensively in clades I, IV and V, which in turn were overlapping similar more ancient events. Similar positive selective pressure acting on plant RGAs has also been reported in other surveys ([15], [34], [30], [11]). In principle, this reservoir of genetic variation could drive the evolution of new R-gene specificities ([30], [31]). To our knowledge this is the first systematically report of evolutionary pressures acting in P. monticola RGAs. Positively selected RGAs branches were not only at terminal positions, implying rather recent positive selective signatures, but also at ancestral positions on the phylogeny. This finding is in accordance with previous reports which showed that numerous selective episodes occur even from the birth of a paralogous locus ([37], [39]).
Signs of positively selected residues were found to be distributed rather evenly across the RGAs amino acid sequences. Positive selection on NBS-containing gene families in plants and especially among their LRR domains, were also reported in various plant hosts ([3], [16], [39], [38], [30], [11]). These findings support the view that selection for durable disease resistance may be a crucial component of plant breeding programs. Additionally, comparative analyses of Rosaceae trees regarding their R genes revealed that solvent-exposed residues of the LRRs domains are hyper-variable with intensive positive selective pressures acting on them ([30]). Moreover, such evidence is consistent with host-pathogen co-evolution processes leading to the acquisition of novel resistance functional specificities ([39], [30]). These findings would facilitate the development of informative RGA-derived molecular markers for identifying fungal resistant P. monticola and P. monticola × P. peuce genotypes in breeding programs using segregating progenies. They may also prove to be of importance for P. peuce itself, as despite the fact that Balkan white pine is a resistant species, future selection pressures on C. ribicola in light of climatic change may alter the virulence of the fungus towards its host.
Conclusions
The P. monticola RGAs gene homologues encoding for R gene members of the TIR-NBS-LRR subfamily were found to be subjected to intensive positive selection, acting widely in three clades across their phylogeny, both on terminal and ancestral lineages. The majority of positively selected residues sites were found to be localized widely across these RGAs sequences, putatively affecting the structures of their ligand-binding domains. Practical applications in the development of disease resistant plants through breeding applications may also be considered, facilitating for instance, the development of informative RGAs-derived sequence characterized amplified region (SCAR) markers, based on RGAs sequences with extensive positive selection acting upon them. Such markers might be particularly useful for identifying fungal resistant genotypes in routine marker-assisted selection (MAS) breeding programs.
List of Abbreviations
The following abbreviations have been used throughout the paper:
- CC: N-terminal coiled-coil domain;
- LRR: Leucine Rich Repeat;
- NBS: nucleotide-binding site domain;
- PAML: Phylogenetic Analysis by Maximum Likelihood;
- RaxML: Randomized Axelerated Maximum Likelihood;
- RGA: Resistance Gene Analog;
- TIR: Toll/interleukin-1 receptor domain;
- WWP: Western white pine.
Acknowledgments
The co-authors have not a conflict of interest to declare.
References
CrossRef | Gscholar
CrossRef | Gscholar
Gscholar
Gscholar
Gscholar
Supplementary Material
Authors’ Info
Authors’ Affiliation
Athanasios Tsaftaris
Laboratory of Genetics and Plant Breeding, Faculty of Agriculture, Forestry & Natural Environment, Aristotle University of Thessaloniki, P.O. Box 261, Thessaloniki GR54124 (Greece)
Aglaea Papadima
Evaggelos Barbas
Filippos A Aravanopoulos
Laboratory of Forest Genetics and Tree Breeding, Faculty of Agriculture, Forestry & Natural Environment, Aristotle University of Thessaloniki, P.O. Box 238, Thessaloniki GR54006 (Greece)
Panagiotis Madesis
Institute of Applied Biosciences, CERTH, Thermi, Thessaloniki, GR57001 (Greece)
Genomedica, Piraeus, GR18537 (Greece)
Corresponding author
Paper Info
Citation
Zambounis A, Avramidou E, Papadima A, Tsaftaris A, Arzimanoglou I, Barbas E, Madesis P, Aravanopoulos FA (2017). Adaptive response of Pinus monticola driven by positive selection upon resistance gene analogs (RGAs) of the TIR-NBS-LRR subfamily. iForest 10: 237-241. - doi: 10.3832/ifor2050-009
Academic Editor
Alberto Santini
Paper history
Received: Mar 09, 2016
Accepted: Sep 27, 2016
First online: Feb 02, 2017
Publication Date: Feb 28, 2017
Publication Time: 4.27 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2017
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 51419
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 43126
Abstract Page Views: 3528
PDF Downloads: 3458
Citation/Reference Downloads: 76
XML Downloads: 1231
Web Metrics
Days since publication: 3431
Overall contacts: 51419
Avg. contacts per week: 104.91
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Mar 2025)
(No citations were found up to date. Please come back later)
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Characterization of two poplar homologs of the GRAS/SCL gene, which encodes a transcription factor putatively associated with salt tolerance
vol. 8, pp. 780-785 (online: 19 May 2015)
Review Papers
Prospects for evolution in European tree breeding
vol. 17, pp. 45-58 (online: 06 March 2024)
Research Articles
Early flowering and genetic containment studies in transgenic poplar
vol. 5, pp. 138-146 (online: 13 June 2012)
Research Articles
Genetic variation and heritability estimates of Ulmus minor and Ulmus pumila hybrids for budburst, growth and tolerance to Ophiostoma novo-ulmi
vol. 8, pp. 422-430 (online: 15 December 2014)
Technical Advances
Gene flow in poplar - experiments, analysis and modeling to prevent transgene outcrossing
vol. 5, pp. 147-152 (online: 13 June 2012)
Review Papers
Indicators of drought effects in Pinus sylvestris: genetic analyses to corroborate the results of empirical methods
vol. 3, pp. 89-91 (online: 15 July 2010)
Research Articles
Preliminary study on genetic variation of growth traits and wood properties and superior clones selection of Populus ussuriensis Kom.
vol. 12, pp. 459-466 (online: 29 September 2019)
Review Papers
Genetic diversity and forest reproductive material - from seed source selection to planting
vol. 9, pp. 801-812 (online: 13 June 2016)
Research Articles
Genetic variation of Fraxinus excelsior half-sib families in response to ash dieback disease following simulated spring frost and summer drought treatments
vol. 9, pp. 12-22 (online: 08 September 2015)
Research Articles
Chloroplast DNA barcoding genes matK and psbA-trnH are not suitable for species identification and phylogenetic analyses in closely related pines
vol. 15, pp. 141-147 (online: 25 April 2022)
iForest Database Search
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords







