
Chloroplast microsatellites as a tool for phylogeographic studies: the case of white oaks in Poland
iForest - Biogeosciences and Forestry, Volume 8, Issue 6, Pages 765-771 (2015)
doi: https://doi.org/10.3832/ifor1597-008
Published: Jul 19, 2015 - Copyright © 2015 SISEF
Research Articles
Abstract
Assessing the distribution of chloroplast DNA (cpDNA) haplotype variation is useful for studying the phylogeography of angiosperms. In the last two decades the cpDNA phylogeography of white oaks in Europe has been extensively studied, mostly based on the PCR-RFLP technique. However, PCR-RFLPs have low mutation rates and are primarily useful for reconstructing patterns at large geographical scales and lack resolution at fine spatial scales. Here we evaluate the usefulness of chloroplast microsatellites (cpSSR) as an alternative to PCR-RFLPs in Polish oak populations which have been underrepresented in previous studies. Eighty-five cpSSR haplotypes were detected using 14 cpSSR loci and a broad collection of 6680 trees sampled throughout Poland. Haplotype diversity was significantly lower in Q. petraea (He = 0.798) than in Q. robur (He = 0.820). Only 17 haplotypes (H01-H17) were found in 13 or more individuals, comprising together 97.9% of the sample. Most frequent cpSSR haplotypes were related to PCR-RFLP haplotypes, establishing the cross-references between the two marker systems. There was significant concordance between the matrices of genetic distances obtained by PCR-RFLP haplotypes and cpSSR haplotypes. Phylogenetic relationships among cpSSR haplotypes supported the existence of the three predominant maternal lineages of oaks in Poland: Iberian (7.8%), Apennine (20.6%) and Balkan (65.5%). The results are discussed with regards to the usefulness of cpSSR markers for phylogeographic studies.
Keywords
Chloroplast Microsatellites, PCR-RFLP, White Oaks, Phylogeography
Introduction
Studies of genetic diversity of chloroplast DNA (cpDNA) can provide important insights into phylogenetic and historical relationships among populations of angiosperms ([22], [35]). For example, the geographic distribution of intraspecific cpDNA variants has been widely used to define post-glacial migration routes and to identify zones of secondary contact ([22], [33]). Additionally, cpDNA has found a practical application in marker-based tracking of forest reproductive material and timber ([5]). Both of these aspects are important for management and the conservation of forest genetic resources ([29]).
The most widespread species of European white oaks, pedunculate oak (Quercus robur L.) and sessile oak (Quercus petraea [Matt.] Libel.) grow naturally across a large part of Europe and are important components of forest ecosystems, with high economic value. The application of the PCR-RFLP technique has allowed the detection of multiple cpDNA haplotypes with specific geographic distributions across Europe, grouped into three major lineages originating from the locations of the primary glacial refugia (Iberia, Apennines, Balkans ([11], [2], [15], [24], [30], [33], [34], [35], [3], [16], [27]). However, these studies focused mostly on Western or Southern European populations, and populations from Central and Eastern Europe (Poland, Baltic Countries, Slovakia and Czech Republic) have received relatively less attention ([4], [40]). Within the Baltic region (54 locations from Estonia, Latvia, Lithuania and Poland) Csaikl et al. ([4]) found 13 haplotypes. On the other hand, Dering et al. ([8]), studying 78 locations in Poland (each consisting of 1 to 12 individuals) detected only 6 PCR-RFLP haplotypes representing the three major maternal lineages. Neither of these studies uniformly covered the distribution of both oak species in Poland, introducing a possible bias to the observed distribution of cpDNA variation in the region.
CpDNA variation can be alternatively mapped with microsatellite sequences (cpSSR - [7]). Several universal cpSSR loci have been identified for angiosperm taxa ([42]). In addition, a number of loci have been designed specifically for oaks ([6]) or in general for Fagaceae ([38]). Since then, several phylogeographic studies have been done in oaks employing cpSSR markers ([21], [26], [32], [39]). Microsatellites differ from PCR-RFLP markers because of their mutation mechanism and rate ([14]). Therefore, they may reveal incomplete concordance with PCR-RFLP when studying the genetic structure of populations. Nonetheless, direct relationships between cpSSR and PCR-RFLP haplotypes is expected since they are fully linked in the chloroplast genome, as previously reported ([7], [17], [29]). However, PCR-RFLPs have low mutation rates and are primarily useful for reconstructing patterns at large geographical scales, while they lack the resolution needed for fine spatial scales studies.
Laboratory techniques for analyses of cpSSR markers are less demanding than for PCR-RFLP variants. CpSSRs, being usually amplified as short DNA fragments, are easily detected even in partially degraded DNA, and they are less sensitive to inhibitory substances present in plant tissues ([16]). They are relatively cost-effective and are easily multiplexed for high-throughput studies. However, cpSSR markers seem to be more sensitive to size homoplasy ([14], [41]), which could complicate phylogenetic inferences. However, the level of homoplasy within species is usually considered low enough to allow population studies ([28]).
Here, we assembled a large collection of Q. petraea and Q. robur trees (n=6680) obtained by different sampling schemes to achieve a representative coverage of the study region. Such a large sample enables the identification of cpSSR haplotype variants existing in Central Europe and can provide a reference for future studies. Therefore, in this paper the identification of polymorphic cpSSR loci within the study region was used to assess the level of genetic diversity in species and populations of oaks. We detected numerous multilocus haplotypes whose relatedness was examined. Additionally, we related the most frequent cpSSR haplotypes to the haplotypes identified in previous studies based on the PCR-RFLP technique. The protocol for an optimized high-throughput genotyping procedure is described for future research.
Material and methods
Plant material
A total of 6680 oak trees were sampled throughout Poland (Fig. S1 in Appendix 1). The samples included individuals from various sampling schemes: 2512 trees from certified seed stands (approx. 20 individuals from each of 132 forest stands), a set of 599 plus trees (phenotypically selected trees utilized in forest tree improvement programs), a separate collection of 3310 trees consisting of up to 5 individuals per species from 420 forest districts where the species were present (uniformly scattered sample), and a subset of 259 old trees (usually older than 250 years) growing individually or in road alleys, some being considered as national nature monuments. Overall, the collection comprised 3938 individuals of Quercus robur and 2742 individuals of Q. petraea. The species status of individuals was determined based on leaf morphology at the time of sampling, and whenever possible (e.g., for plus trees and seed stands) also verified in the State Forest Reproductive Material Office.
DNA isolation
Total genomic DNA was extracted from 50 mg of dried leaves. Plant material was ground in a Mixer Mill MM301 (Retsch, Haan, Germany). DNA isolation followed a CTAB protocol ([9]). The amount and quality of DNA was evaluated and adjusted to a concentration of 10 ng/µl using DNA calculator (Biophotometer®, Eppendorf, Hamburg, Germany).
Chloroplast microsatellites
For the initial screening of polymorphism at cpSSR loci we used a representative subsample of individuals consisting of DNA from 282 plus trees (198 Q. robur, 84 Q. petraea) from 64 Forest Districts in Poland, widely distributed across the country. We selected 35 cpSSR loci that had been used in population genetic studies of forest trees, especially in oaks (see Table S1 in Appendix 1), including 8 markers developed for dicotyledonous angiosperms ([42]), 16 made specifically for oaks ([6]) and 11 created for Fagaceae ([38]). The loci were divided into four marker sets, each consisting of 6-11 loci, and subjected to PCR-multiplex protocols (Table S1 in Appendix 1). However, among the 35 pre-selected cpSSR loci only 14 (ccmp4, cmcs12, cmcs5, cmcs6, cmcs7, cmcs8, cmcs9, µcd1, µcd4, µcd5, µdt1, µdt3, µdt4, µkk4) were polymorphic in the test subsample. The set of 14 loci was subjected to PCR-multiplex optimization in order to establish a single PCR reaction. Finally the optimized reaction mix contained: 1x PCR Buffer, 1.25 mM MgCl2, 0.25 mM dNTP-Mix, 0.25 U Taq polymerase (Taq PCR Core Kit®, Qiagen, Hilden, Germany), 0.5 mg/ml BSA, 0.2 µM µcd4, 0.125µM µdt3, 0.075µM cmcs8, 0.15µM cmcs9, 0.1µM ccmp4, 0.125µM cmcs7, 0.05µM µcd5, 0.05µM µcd1, 0.125µM cmcs5, 0.075µM cmcs12, 0.05µM µdt1, 0.15µM µkk4, 0.25µM µdt4, 0.15µM cmcs6 (concentration refers to that of both primers) and 10 ng of template DNA. Primers from these loci were fluorescently labeled (see Table S1 in Appendix 1). PCR conditions were as follows: an initial denaturation step at 95°C for 5 min, followed by 8 touchdown cycles at 94°C for 30 s, 1:30 s at 56 °C (-1 °C/cycle), 1 min at 72 °C, 24 cycles at 94 °C for 30 s, 45 s at 48 °C, 1 min at 72 °C, and a final extension at 72 °C for 10 min. The amplification products were separated using an ABI 3130XL sequencer (Applied Biosystems, Foster City, CA, USA), with LIZ600 as an internal size standard. The identification of alleles based on their size was determined using GENMAPPER® software v. 4.0 (Applied Biosystems). The accuracy of genotyping based on the 14-plex PCR reaction was verified for 16 randomly chosen individuals, by repeating single PCR reactions of each locus. This indicated full concordance of allele sizes determined in multiplex and singleplex reactions. Finally, the 14-plex PCR protocol was used to analyze all 6680 samples.
PCR-RFLP haplotypes
PCR-RFLP haplotypes were also determined on a subset of 282 individuals in order to relate cpSSR haplotypes to the PCR-RFLP haplotypes identified in earlier studies ([4], [34], [8]). PCR-RFLP variants were assayed following the protocol described by Dering et al. ([8]) and references therein. The interpretation of the restriction pattern followed Petit et al. ([34]).
Data analysis
For analysis of cpSSR data, each species was treated as single population. Allelic frequencies, number of alleles and genetic diversity were calculated using the FSTAT software ([20]). Haplotypes were determined as a combination of different microsatellite variants across the cpSSR loci. We used the software HAPLOTYPE ANALYSIS© ver. 1.05 ([13]) to estimate haplotype frequencies and the following genetic diversity measures: number of haplotypes (A), effective number of haplotypes (Ne), haplotypic richness (HR - [12]), genetic diversity (He), and the mean genetic distance between individuals (Dsh2 - [18]).
The phylogeny of the established cpSSR haplotypes was inferred based on the median-joining method ([1]). Maximum-parsimony analysis was conducted using the software NETWORK ver. 4.6.1.2 (Fluxus Technology Ltd, ⇒ http://⇒ www.fluxus-engineering.com). Additionally, phylogenetic relationships were assessed based on genetic distances between individual haplotypes ([18]), using the neighbor-joining algorithm, implemented in the software package TREEVIEW ([31]).
After establishing the correspondence between PCR-RFLP and cpSSR haplotypes based on the subset of 282 plus trees, the matrix of genetic distances between PCR-RFLP haplotypes presented in Kremer et al. ([25]), was compared to the matrix of genetic distances between cpSSR haplotypes ([18]) using a Mantel test implemented in the R package “ecodist” ([19]). The significance of the Mantel test was evaluated with 100 000 random permutations.
Results
Genetic diversity
Overall, we detected 45 alleles among 6680 oak individuals based on 14 cpSSR loci. The number of alleles per locus ranged from 2 (cmcs7, µcd1, µcd5, µkk4) to 5 (cmcs6 - Tab. 1), with an average of 3.214 ± 0.261 (SE). Genetic diversity of individual loci ranged from 0.001 to 0.525, with a mean of 0.281 ± 0.052. Although the number of alleles found was greater in Q. robur, the genetic diversity was generally higher in Q. petraea (Tab. 1). Genetic diversity at individual loci revealed high interspecific consistency (r=0.946; p<0.001), indicating that these loci can be equally informative about genetic diversity in both species.
Tab. 1 - Genetic diversity of cpSSR loci based on Quercus robur and Q. petraea. (A): number of alleles; (He): genetic diversity; (SE): standard error.
| Locus | Quercus robur (n=3938) |
Quercus petraea (n=2742) |
Both species (n=6680) |
|||
|---|---|---|---|---|---|---|
| A | H e | A | H e | A | H e | |
| cmcs12 | 3 | 0.002 | 1 | 0.000 | 3 | 0.001 |
| cmcs5 | 3 | 0.300 | 3 | 0.401 | 3 | 0.346 |
| cmcs6 | 5 | 0.399 | 5 | 0.462 | 5 | 0.429 |
| cmcs7 | 2 | 0.003 | 1 | 0.000 | 2 | 0.001 |
| cmcs8 | 3 | 0.028 | 3 | 0.032 | 3 | 0.030 |
| µdt1 | 4 | 0.405 | 4 | 0.445 | 4 | 0.421 |
| µdt3 | 4 | 0.447 | 4 | 0.494 | 4 | 0.472 |
| µdt4 | 4 | 0.394 | 3 | 0.523 | 4 | 0.454 |
| ccmp4 | 3 | 0.299 | 2 | 0.175 | 3 | 0.252 |
| cmcs9 | 4 | 0.395 | 3 | 0.523 | 4 | 0.454 |
| µcd1 | 2 | 0.083 | 2 | 0.018 | 2 | 0.057 |
| µcd4 | 4 | 0.525 | 3 | 0.506 | 4 | 0.525 |
| µcd5 | 2 | 0.138 | 2 | 0.183 | 2 | 0.157 |
| µkk4 | 2 | 0.278 | 2 | 0.395 | 2 | 0.331 |
| Mean | 3.214 | 0.264 | 2.714 | 0.296 | 3.214 | 0.281 |
| SE | 0.261 | 0.048 | 0.304 | 0.058 | 0.261 | 0.052 |
Haplotype diversity
The use of 14 cpSSR loci resulted in the detection of 85 different haplotypes across both species. The number of haplotypes was higher in Q. robur (67) than Q. petraea (47 - Tab. 2). Haplotypes were ranked based on their observed frequencies across both species and labels (H01, H02, etc.) were assigned according to their frequency in descending order, i.e., starting from the most frequent haplotype. The frequency spectrum of the haplotypes was highly skewed, with H49-H85 found in just one individual, whilst each of H34-H48 occurred in just two. Seventeen haplotypes (H01-H17) were found in 13 or more individuals; 97.9% of the collection had one of these 17 haplotypes. Frequency dropped off rapidly for haplotypes lower than H10 in the ranking. Full details of all multilocus haplotypes are presented in Appendix 1 (Table S2). The 17 most frequent haplotypes could be characterized using a subset of 5 cpSSR loci only, as shown in Tab. 3. Some 25 haplotypes were shared by both species, with 39 and 21 (mostly rare haplotypes) found exclusively in Q. robur and Q. petraea, respectively. The effective number of haplotypes was 5.543 and 4.936 in Q. robur and Q. petraea, respectively, with a mean of 5.367 estimated across both species. The effective number of haplotypes was significantly lower in Q. petraea than in Q. robur (p < 0.001 after permutation test with individual haplotypes taken as units). Haplotype diversity was lower in Q. petraea (He = 0.798) than in Q. robur (He = 0.820 - p < 0.001 after permutation test). The mean genetic distances between individuals within species (Dsh2) were comparable (Tab. 2).
Tab. 2 - Measures of haplotypic diversity for Q. robur and Q. petraea based on 14 cpSSR loci, and the subset of 5 loci (ccmp4, cmcs5, cmcs6, µcd4, µdt1; see the text).
| Parameter |
Quercus
robur |
Quercus
petraea |
Both species |
Both species (subset of 5 loci) |
|---|---|---|---|---|
| Number of individuals (N) | 3938 | 2742 | 6680 | 6680 |
| Number of haplotypes (A) | 67 | 47 | 85 | 43 |
| Effective number of haplotypes (Ne) | 5.543 | 4.936 | 5.367 | 5.267 |
| Genetic diversity (He) | 0.820 | 0.798 | 0.814 | 0.810 |
| Mean genetic distance between individuals (Dsh2) | 2.589 | 2.629 | 2.618 | 1.924 |
Tab. 3 - Genotypes of the most frequent haplotypes based on the subset of 5 loci (ccmp4, cmcs5, cmcs6, µcd4, µdt1;). Allele names are given based on their size (bp).
| Haplotype | ccmp4 | cmcs5 | cmcs6 | µcd4 | µdt1 | Count | Freq |
|---|---|---|---|---|---|---|---|
| H01 | 147 | 200 | 79 | 114 | 96 | 2219 | 0.3281 |
| H02 | 146 | 198 | 79 | 114 | 97 | 1179 | 0.1749 |
| H03 | 147 | 200 | 78 | 114 | 97 | 1169 | 0.1731 |
| H04 | 147 | 200 | 79 | 115 | 96 | 767 | 0.1135 |
| H05 | 147 | 200 | 81 | 114 | 97 | 302 | 0.0442 |
| H06 | 147 | 197 | 79 | 114 | 96 | 245 | 0.0366 |
| H07 | 146 | 198 | 79 | 115 | 95 | 176 | 0.0261 |
| H08 | 147 | 200 | 80 | 114 | 97 | 168 | 0.0245 |
| H09 | 147 | 201 | 79 | 114 | 96 | 124 | 0.0178 |
| H10 | 147 | 200 | 79 | 114 | 97 | 109 | 0.0154 |
| H11 | 147 | 199 | 80 | 114 | 97 | 59 | 0.0088 |
| H12 | 148 | 200 | 79 | 114 | 96 | 28 | 0.0037 |
| H13 | 147 | 200 | 78 | 114 | 96 | 22 | 0.0027 |
| H14 | 146 | 200 | 79 | 114 | 96 | 18 | 0.0025 |
| H15 | 146 | 197 | 79 | 115 | 97 | 18 | 0.0024 |
| H16 | 146 | 198 | 79 | 114 | 96 | 16 | 0.0024 |
| H17 | 146 | 200 | 78 | 114 | 97 | 13 | 0.0019 |
| Sum | 6632 | 0.9904 | |||||
Relationship between PCR-RFLP and cpSSR haplotypes
Haplotypes based on cpSSR loci were compared to the PCR-RFLP haplotypes in the subset of 282 trees. Six different PCR-RFLP haplotypes belonging to the three major maternal lineages (Apennine, Iberian, Balkan) were found, with each haplotype present at least in 3 individuals (Tab. 4). The cpSSR haplotypes H02 and H07 could be assigned to the PCR-RFLP haplotypes RFLP1 and RFLP2, respectively, both belonging to the Apennine lineage C ([34]). The haplotype H05 represents most likely the haplotype RFLP12, which is part of the Iberian maternal lineage B. Finally, the most frequent Balkan maternal lineage A consisted of three RFLP haplotypes: RFLP4 (related mostly to cpSSR haplotypes H03 and H06), RFLP7 (H01) and RFLP5 (related mostly to H04, H06, H10 - Tab. 4). Notably, while RFLP1, RFLP2 and RFLP12 haplotypes were connected in general to single cpSSR haplotypes, the Balkan PCR-RFLP haplotypes were related to several different cpSSR haplotypes. In particular, haplotype RFLP5 appeared highly heterogeneous and was related to nine different cpSSR haplotypes, which included quite divergent haplotypes such as H04, H06, H08, H09, H10 and H12 (the effective number of cpSSR haplotypes within RFLP5 haplotype was 2.889 - see Tab. 4). The spatial distribution of PCR-RFLP haplotypes generally followed the distributions observed in earlier studies ([4], [8], Chmielewski et al., in preparation).
Tab. 4 - Number of individuals with specific combinations of PCR-RFLP and cpSSR haplotypes. (*): indicate the most frequent PCR-RFLP haplotype within a given cpSSR haplotype. (AEcpSSR): effective number of PCR-RFLP haplotypes within a given cpSSR haplotype; (AERFLP): effective number of cpSSR haplotypes within a given PCR-RFLP haplotype.
| Haplo | RFLP1 | RFLP2 | RFLP4 | RFLP5 | RFLP7 | RFLP12 | Count | AE cpSSR |
|---|---|---|---|---|---|---|---|---|
| H01 | - | - | 1 | 3 | 95* | - | 99 | 1.085 |
| H02 | 42* | - | - | - | - | - | 42 | 1.000 |
| H03 | 1 | - | 31* | - | - | - | 32 | 1.064 |
| H04 | - | - | - | 51* | 1 | - | 52 | 1.039 |
| H05 | - | - | - | 1 | - | 3* | 4 | 1.600 |
| H06 | - | - | 5 | 11* | - | - | 16 | 1.753 |
| H07 | 1 | 7 | 1 | - | - | - | 9 | 1.588 |
| H08 | - | - | - | 3* | 1 | - | 4 | 1.600 |
| H09 | - | - | - | 5* | - | - | 5 | 1.000 |
| H10 | - | - | - | 12* | - | - | 12 | 1.000 |
| H12 | - | - | 1 | 2* | - | - | 3 | 1.800 |
| H25 | - | - | - | 4* | - | - | 4 | 1.000 |
| Count | 44 | 7 | 39 | 92 | 97 | 3 | 282 | - |
| AE RFLP | 1.096 | 1.000 | 1.538 | 2.889 | 1.042 | 1.000 | - | - |
We used a Mantel test to compare the matrix of genetic distances between PCR-RFLP haplotypes presented in Kremer et al. ([25]) to the matrix of genetic distances between cpSSR haplotypes related to the respective PCR-RFLP haplotypes, as established in this study based on Goldstein et al. ([18]), e.g., the distance between RFLP1 and RFLP7 was compared to the distance between H01 and H02 (see Tab. 4). The test revealed a significant correlation between the two types of genetic distances (Pearson’s r = 0.570, p < 0.05), suggesting that phylogenetic relationships estimated by cpSSR haplotypes were fairly similar to those obtained by PCR-RFLP haplotypes.
Phylogenetic relationship among cpSSR haplotypes
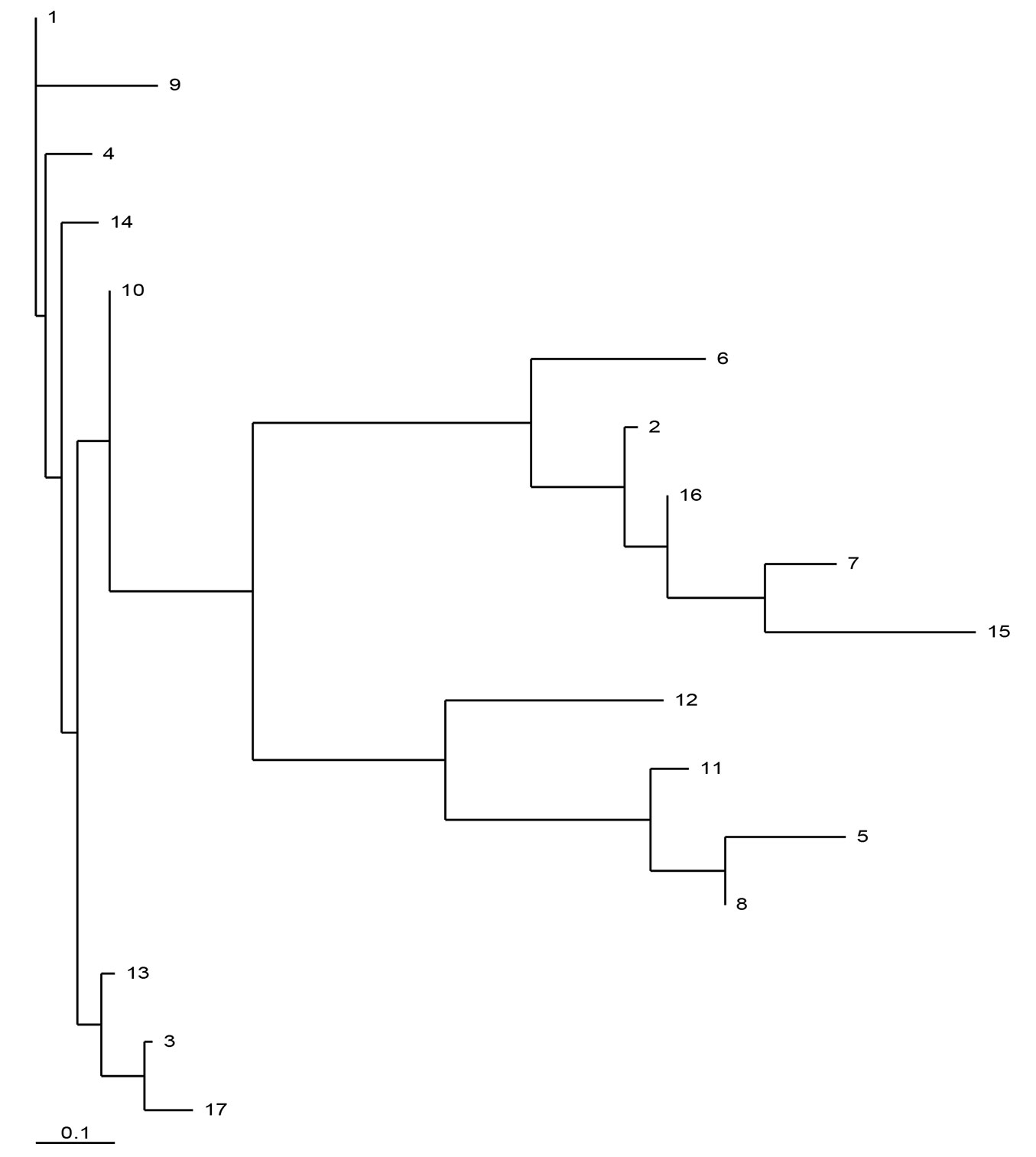
The maximum parsimony haplotype network based on 14 cpSSR loci for the 17 most frequent haplotypes (n≥13) is presented in Fig. 1. The haplotypes clustered in three major clades. The largest and centrally located group includes 8 haplotypes: H01, H03, H04, H09, H10, H13, H14, H17, with a cumulative frequency of 65.5%. This group is related to the Balkan maternal lineage A, based on the above-mentioned relationships between PCR-RFLP and cpSSR variants. Another group consisted of 4 haplotypes H02, H07, H15 and H16 (cumulative frequency: 20.6%) representing the Apennine maternal lineage C; while 3 haplotypes, H05, H08 and H11 (cumulative frequency: 7.8%) were assigned to the Iberian lineage B. The haplotypes H06 and H12 seem to be intermediate between the lineages A and C and between lineages A and B, respectively (Fig. 1). The inferred phylogenetic relationships between haplotypes were confirmed when measured based on genetic distances ([18]) using the neighbor-joining algorithm (Fig. 2).
Fig. 1 - Maximum parsimony phylogenetic network among the 17 most frequent haplotypes (n ≥ 13) for 14 cpSSR loci. Circle sizes are proportional to the haplotype frequency. Red dots (median vectors) and black dots represent missing or not sampled haplotypes.
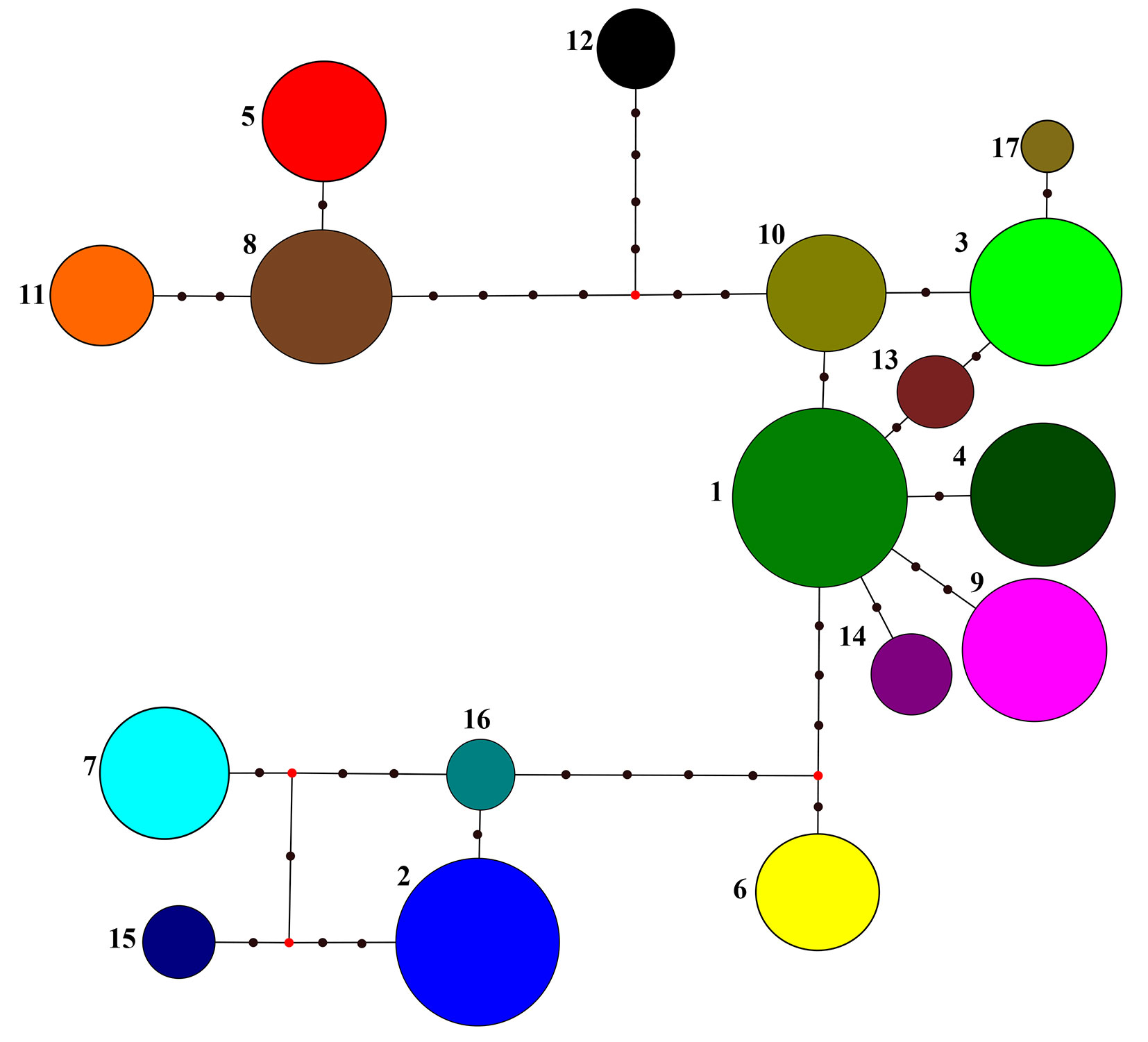
Fig. 2 - Neighbor-joining tree of cpDNA haplotypes for the 17 most frequent haplotypes (n≥13 ) based on pairwise genetic distances ([18]) estimated from 14 cpSSR loci (2000 bootstraps).
Each of the rare cpSSR haplotypes (H18-H85) might be related to one of the most frequent haplotypes (H1-H17 - Fig. S2 in Appendix 1). When we assign all haplotypes to one of five major haplotype groups (concentrated around most frequent haplotype lineages H01, H02, H05, H06, H12), the number of different haplotypes within each group is proportional to the number of individuals observed within haplogroups (r = 0.978, p = 0.004). Similarly, when we consider nine haplogroups (given that the largest major haplotype lineage is divided into smaller haplogroups H01, H03, H04, H09, H10), the number of haplotypes within such haplogroups is still proportional to the number of individuals observed within haplogroups (r = 0.772, p = 0.015). This suggests that the number of different haplotypes is related to the overall sample size of a haplogroup.
Optimization
The use of all 14 cpSSR loci resulted in the detection of 85 different haplotypes over the two species analyzed. However, as mentioned above, the majority of haplotypes were found in just one or two individuals, and only 17 haplotypes (H01-H17) were found in 13 or more individuals. Therefore, we optimized the genotyping procedure in order to minimize the number of cpSSR loci to be used for screening, whilst still being able to detect the 17 most frequent haplotypes. We tested various combinations of loci and found that the 17 most frequent haplotypes could be characterized using just 5 loci (ccmp4, cmcs5, cmcs6, µcd4, µdt1). With these five loci we were able to detect 43 different haplotypes (Tab. 3). The optimized set of loci will be valuable for future research and for practical, marker-based forest management (e.g., tracking of forest reproductive material or gene conservation).
Discussion
Studies using cpSSR markers have usually reported higher levels of genetic variation than studies based on the PCR-RFLP technique ([36]). Chloroplast microsatellites have already been used for studying phylogeography in oaks ([21], [26], [32]), revealing patterns of variation similar to PCR-RFLP markers ([7], [16]). However, recent studies of cpSSR diversity in European white oaks based on chloroplast polymorphisms indicated a smaller number of haplotypes compared with this study ([21], [29]).
In this paper, 14 cpSSR loci were analyzed to identify 85 distinct haplotypes on a large number of individuals sampled across Poland. Such a large number of cpSSR haplotypes contrasts with the amount of PCR-RFLP haplotypes found earlier in the region ([4], [8]). However, the majority of haplotypes were rare, with frequencies below 0.001. The number of cpSSR haplotypes was larger for Q. robur than for Q. petraea (67 vs. 47, respectively), reflecting the overall trend found across Europe based on PCR-RFLP markers (23 vs. 17 - [34]). Sharing of haplotypes between the two oak species confirms their close phylogenetic relationship and/or ability to hybridize ([11], [34]).
To date, only a few studies have attempted to relate chloroplast haplotypes based on cpSSRs and PCR-RFLP markers. Defining such relatedness is necessary if new phylogeographic studies based on cpSSRs are to be compared to earlier works carried out using PCR-RFLPs. Deguilloux et al. ([7]) found almost complete redundancy between the haplotypes identified using the two types of markers in French oak populations. Other studies related haplotypes from the two marker systems only partially ([17], [29]).
The analysis of the phylogenetic relationships among the 17 most frequent haplotypes (n>13) and their associations to PCR-RFLP haplotypes confirmed the existence of the three major clades related to the main maternal lineages previously described in this area ([33], [4], [8]). The most frequent one appeared to be the Balkan maternal lineage A (nearly 64%), followed by the Apennine lineage C (21%) and the Iberian lineage B (8%). The cpSSR haplotypes H06 and H12 were found to be intermediate between different clades (H06 intermediate between Balkan and Apennine, H12 intermediate between Balkan and Iberian lineages - Fig. 1), but both were assigned to haplotype RFLP5 which represents the Balkan lineage. While PCR-RFLP haplotypes of the Apennine and Iberian lineages were largely monotypic as for the number of cpSSR haplotypes found, the Balkan lineage included several cpSSR haplotypes. In particular, haplotype RFLP5 appeared highly heterogeneous as it was related to seven cpSSR haplotypes. Previously, Petit et al. ([34]) and Bordács et al. ([2]) suggested that this haplotype might have persisted in both the Apennine and Balkan refugia. In Slovakia, Tutkova-van Loo & Burg ([40]) found that haplotype RFLP5 could be resolved as three related variants (5A, 5B, and 5C).
We have found a large number of rare haplotypes (with frequency < 0.001), which were the result of mutations in one of the 14 cpSSR loci. Petit et al. ([34]) recorded numerous rare PCR-RFLP haplotypes; however, these haplotypes were not confirmed by the reference laboratory. Rare haplotypes are generally of minor importance in phylogeographic studies, but they might be useful for identifying specific populations when showing an increased local frequency. An assessment of the spatial distribution of rare haplotypes (local or widely dispersed) might provide insights into the spread dynamics of new cpDNA variants.
The high mutation rates of cpSSR regions increase the potential for homoplasy. This may cause problems in phylogenetic studies, especially when performed at a very large spatial scale ([10], [37], [14]). However, such problem should be negligible for studies at small geographical scales. Furthermore, homoplasy level within species is usually considered as low enough to allow population genetic analysis ([28]). In our study, we found similar relatedness patterns among PCR-RFLP and cpSSR haplotypes. A given cpSSR haplotype was related mostly to a single dominant PCR-RFLP haplotype, suggesting that our data set was affected by homoplasy to a limited degree. Similarly, a given PCR-RFLP haplotype was mostly related to a single dominant cpSSR haplotype, indicating some cryptic diversity within PCR-RFLP haplotypes. A distinct exception, haplotype RFLP5 (see above), needs further investigation.
The number of distinct haplotypes detected in the study area is expected to depend on two main factors. Firstly, it should be related to the effective size of a migrating population. Secondly, it may depend on the migration distance from the refugium. As a result of long-distance migration, when effective population size decreases dramatically (bottleneck), rare haplotypes tend be lost. On the other hand, new haplotypes might be generated through mutation. The theory predicts that the number of different variants in a sample is a function of the effective size and the mutation rate ([23]). Assuming that mutation rate is constant for a given locus, the number of haplotypes in our sample should be determined mainly by the effective population size. As a consequence, the prevalence of specific haplotypes centered on haplotype H01 can be explained by historically-determined, unbalanced representation of specific maternal lineages (refugia). It should be noticed that our sampling was fairly uniform across the country (Fig. S1 in Appendix 1), thus the number of haplotypes within the maternal lineages was not affected by the clustered distribution of lineages ([8]).
We found that as few as five cpSSR loci were necessary to characterize most frequent haplotypes in white oaks, indicating some redundancy among loci. This set of five loci could serve as a basis for standard assessment of haplotypic diversity for gene conservation or forest reproductive material (FRM) traceability surveys. However, using the established multiplex of 14 loci may be helpful in detecting new local haplotypes, which could be used for genetic fingerprinting of local FRM or timber.
Conclusions
Chloroplast microsatellites, with their amenability to multiplex PCR and automation can be considered markers of choice for population genetic studies in white oaks. Our study confirmed the relationship between haplotypes established with cpSSR and PCR-RFLP polymorphisms, making possible comparisons with previous studies. CpSSR markers have the potential to detect a relatively larger number of haplotypes, which in turn might cause problems in interpretation of results. Nevertheless, occasional rare haplotypes can be easily “binned” with related, more frequent haplotypes to simplify analysis. In-depth analysis of the spatial distribution of cpSSR haplotypes of the two oak species should provide a detailed picture of the current distribution of maternal lineages. This will be helpful in reconstructing the migratory routes of the species and potentially would allow the detection of local population differentiation.
Acknowledgements
We thank Dr. Stephen Cavers and two anonymous reviewers for valuable comments and suggestions, which significantly improved the paper. The study was supported by the Ministry of Science and Higher Education of the Republic of Poland (Grant No: NN309706040). The authors are thankful to Ewa Sztupecka for laboratory assistance, and to Lukasz Kubera and the personnel of State Forests of Poland for their help in collecting oak samples in the field.
MC, JB planned the research and wrote the article. MC, IJC and JB contributed to statistical analyses. JB obtained funding and organized the sample collection. AD organized uniformly scattered sample collection. ML performed PCR-RFLP analyses. MC, KM and IJC optimized cpSSR workflow and designed PCR multiplexes. MC and KM performed cpSSR laboratory analyses.
References
Gscholar
Online | Gscholar
Online | Gscholar
Gscholar
CrossRef | Gscholar
Gscholar
Gscholar
CrossRef | Gscholar
CrossRef | Gscholar
Authors’ Info
Authors’ Affiliation
Katarzyna Meyza
Igor J Chybicki
Artur Dzialuk
Jaroslaw Burczyk
Institute of Experimental Biology, Department of Genetics, Kazimierz Wielki University of Bydgoszcz, Chodkiewicza 30, 85-064 Bydgoszcz (Poland)
Institute of Dendrology, Polish Academy of Sciences, ul. Parkowa 5, 62-035 Kórnik (Poland)
Corresponding author
Paper Info
Citation
Chmielewski M, Meyza K, Chybicki IJ, Dzialuk A, Litkowiec M, Burczyk J (2015). Chloroplast microsatellites as a tool for phylogeographic studies: the case of white oaks in Poland. iForest 8: 765-771. - doi: 10.3832/ifor1597-008
Academic Editor
Andrea Piotti
Paper history
Received: Feb 12, 2015
Accepted: Jun 01, 2015
First online: Jul 19, 2015
Publication Date: Dec 01, 2015
Publication Time: 1.60 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2015
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 55233
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 44008
Abstract Page Views: 5015
PDF Downloads: 4672
Citation/Reference Downloads: 85
XML Downloads: 1453
Web Metrics
Days since publication: 4012
Overall contacts: 55233
Avg. contacts per week: 96.37
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Mar 2025)
Total number of cites (since 2015): 23
Average cites per year: 2.09
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Comparison of range-wide chloroplast microsatellite and needle trait variation patterns in Pinus mugo Turra (dwarf mountain pine)
vol. 10, pp. 250-258 (online: 11 February 2017)
Short Communications
Differentiation of Populus species by chloroplast SNP markers for barcoding and breeding approaches
vol. 8, pp. 544-546 (online: 13 November 2014)
Research Articles
Clonal structure and high genetic diversity at peripheral populations of Sorbus torminalis (L.) Crantz.
vol. 9, pp. 892-900 (online: 29 May 2016)
Research Articles
Seedling emergence capacity and morphological traits are under strong genetic control in the resin tree Pinus oocarpa
vol. 17, pp. 245-251 (online: 16 August 2024)
Research Articles
Genetic variation and heritability estimates of Ulmus minor and Ulmus pumila hybrids for budburst, growth and tolerance to Ophiostoma novo-ulmi
vol. 8, pp. 422-430 (online: 15 December 2014)
Research Articles
Chloroplast DNA barcoding genes matK and psbA-trnH are not suitable for species identification and phylogenetic analyses in closely related pines
vol. 15, pp. 141-147 (online: 25 April 2022)
Research Articles
Patterns of genetic variation in bud flushing of Abies alba populations
vol. 11, pp. 284-290 (online: 13 April 2018)
Research Articles
Genetic diversity of core vs. peripheral Norway spruce native populations at a local scale in Slovenia
vol. 11, pp. 104-110 (online: 31 January 2018)
Review Papers
Genetic diversity and forest reproductive material - from seed source selection to planting
vol. 9, pp. 801-812 (online: 13 June 2016)
Research Articles
Age trends in genetic parameters for growth and quality traits in Abies alba
vol. 9, pp. 954-959 (online: 07 July 2016)
iForest Database Search
Search By Author
Search By Keyword
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords
PubMed Search
Search By Author
Search By Keyword







