Fungal and bacterial communities in a forest relict of Pinus pseudostrobus var. coatepecensis
iForest - Biogeosciences and Forestry, Volume 16, Issue 6, Pages 299-306 (2023)
doi: https://doi.org/10.3832/ifor4284-016
Published: Nov 09, 2023 - Copyright © 2023 SISEF
Research Articles
Abstract
Mexico is a center of diversity for the genus Pinus, with 44% of pine species being endemic to the country. Mexican pine forests are recognized as hotspots for ectomycorrhizal fungi and bacteria due to the extensive interactions that take place between microorganisms and plants in their roots. These microorganisms play a vital role in the survival of pine species. This study aims to identify fungal and bacterial communities in a relict Mexican pine forest and evaluate the influence of soil physicochemical parameters on microbial composition. Sampling was conducted along a 145 m transect in an isolated natural relict of P. pseudostrobus var. coatepecensis, which is located within a commercial plantation of Pinus patula. A total of 18 soil samples were collected at predetermined distances along the transect, with replicated sampling points as follows: six samples at 20 cm intervals, four samples at 1 m intervals, four samples at 10 m intervals, and four samples at 25 m intervals. The results indicate that fungal composition varies even at short distances and is influenced by the C:N ratio, total carbon (C), total phosphorus (P), and total hydrogen ion concentration (H+). Ectomycorrhizal fungi (EcM) exhibited a higher relative abundance compared to saprotrophic and pathogenic fungi. A total of 69 EcM ASVs (Amplicon Sequence Variants) were identified, being the dominant genera Tomentella, Clavulina, Suillus, Russula, and Elaphomyces. Bacterial communities did not show significant variation in relation to the distance from the sampling points, but soil pH was identified as the main factor of bacterial composition. Dominant bacterial genera included Burkholderia, Bryobacter, Acidobacterium, and Acidothermus. Additionally, it was observed that current soil conditions influenced β diversity. Overall, the results demonstrate that soil fungal and bacterial communities associated with P. pseudostrobus exhibit a unique composition compared to other natural forest systems in the Neotropics.
Keywords
Bacteria, Diversity, Soil, Ectomycorrhizal Fungi, Pinus, Plantation
Introduction
Pinus pseudostrobus Lindl., a species known for its high genetic variation, is native to the Neotropical zone ([47]). The majority of its populations are concentrated along the “Eje Neovolcánico Transversal” (Trans-Mexican Volcanic Belt, Central Mexico). Within the species, P. pseudostrobus var. coatepecensis Martínez holds particular importance due to the quality of its wood; however, it remains relatively understudied. Over the past few decades, there has been a noticeable decline in natural populations resulting in the emergence of forest relicts ([1]) in transformed forest or agricultural landscapes. This forest matrix serves as a valuable resource for the macro- and microbiota inhabiting these forest relicts, facilitating their dispersal and ensuring their survival ([12]).
Ectomycorrhizal fungi (EcM) play a crucial ecological role associated with pine roots, contributing to the survival of pines, by enhancing water uptake and nutrient acquisition. They also improve the resistance of host plants to drought, salinity, heavy metals, and pathogens ([8]). These fungi play a fundamental role in the ecosystem functioning, inducing morphological changes at the root level, and expanding root exploration through the development of external mycelium and fungal mantle, which are influenced by bacteria ([37]). The bacterial community responds to changes in host photosynthetic activity, as well as drought conditions and precipitation levels ([37]).
Bacteria can act as growth promoters (PGPR) in the plant rhizosphere and as mycorrhizal helper bacteria (MHB) in association with mycorrhizal plant roots. Bacteria-mycorrhiza interactions play a vital role in organic matter mineralization, nutrient acquisition, carbon (C) dynamics, and nitrogen (N) provision for plants, including biological nitrogen fixation or N-mineralization. This enhances resistance against pathogens and contributes to host survival ([39]). Changes in microbial community richness and diversity are closely linked to available habitat and forestry practices ([9]). Spiesman et al. ([44]) demonstrated that patch isolation and the type of matrix habitat increase bacterial richness and composition. This suggests that suitable feeding, shelter, or climatic conditions can be found across the matrix, allowing the dispersal and survival of biota inhabiting fragments ([25]). Dispersal mechanisms may limit the establishment of EcM fungi, such as host compatibility, spore germination capacity in response to the presence or absence of roots, and low abundance of spore-dispersing mammals or birds ([2]). Consequently, differences in mycorrhizal community assembly can occur between isolated or adjacent stands within different forest matrices. In current landscapes, these natural forest relicts hold significant potential for conserving soil microorganism diversity ([22]). Therefore, the objectives of this study were (i) to characterize fungal and bacterial communities associated with the roots of P. pseudostrobus var. coatepecensis, and (ii) to identify the soil physicochemical factors influencing microbial composition.
Materials and methods
Study area and remnant description
The study was conducted in the central-eastern part of Mexico, specifically in the state of Veracruz (19° 26′ 04.12″ N, 97° 04′ 19.5″ W), at an altitude of 2209 m a.s.l. The study site covered an area of 25 hectares. The climate in this region is classified as humid temperate [C(f)], with mean annual temperatures ranging from 12 to 18 °C. The coldest month experiences temperatures between -3 and 18 °C, while the hottest month remains below 22 °C. The driest month receives more than 40 mm of precipitation, and rainfall occurs throughout the year, with winter rainfall accounting for over 18% of the annual total ([28]).
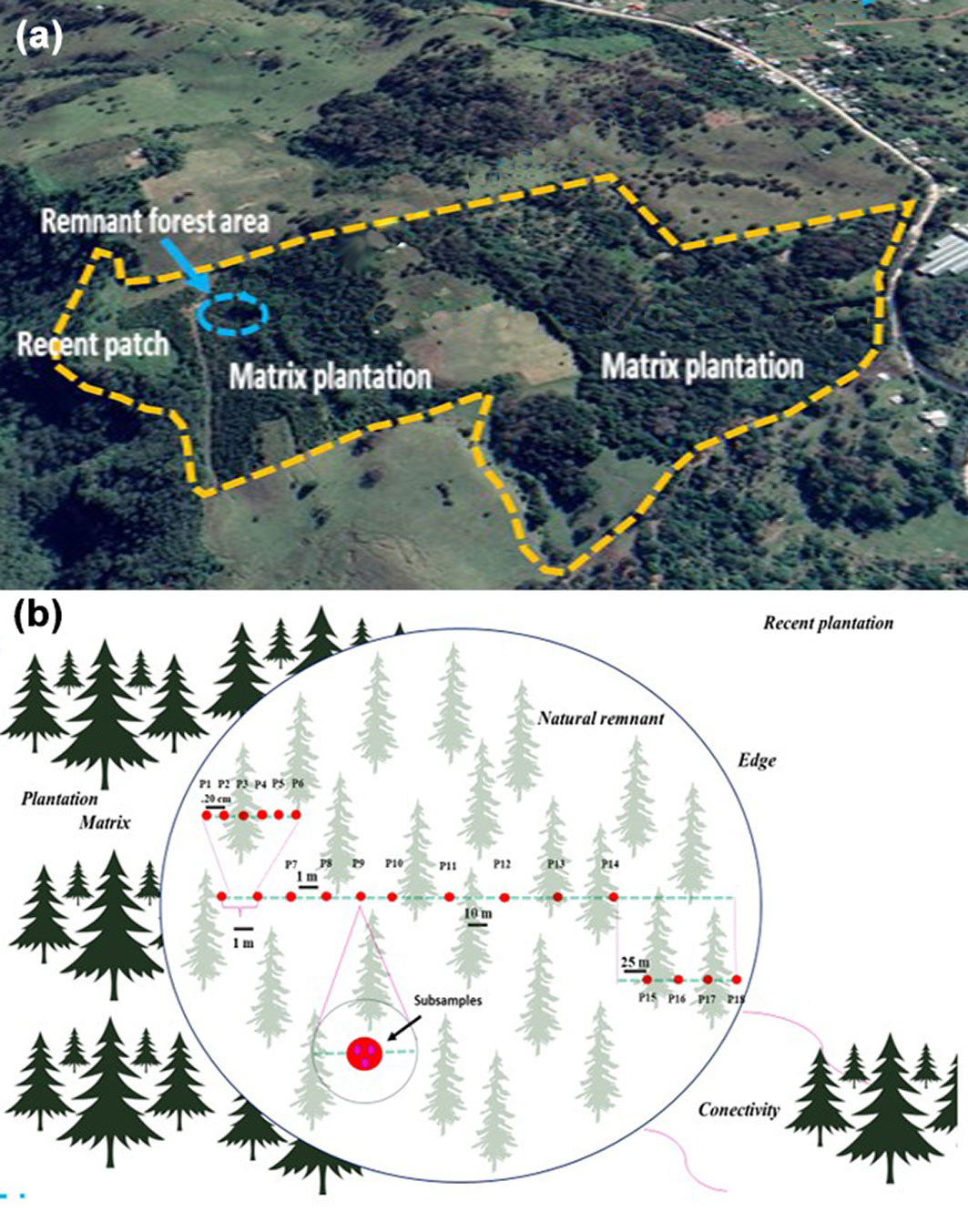
The soil in the study area is primarily Andosol, which develops in volcanic deposits rich in glass under various climatic conditions, except for hyper-arid habitats. However, Andosols can also form on other silicate-rich materials through acid weathering in humid and per-humid climates ([20]). The primary vegetation in the area consists of Pinus patula Schl. et Cham and P. pseudostrobus var. coatepecensis, along with Montane Cloud Forest elements such as Liquidambar styraciflua L., Carpinus caroliniana Thomas Walter, Clethra spp., Cupressus lusitanica Mill., and Quercus spp. The original forest has been fragmented due to agricultural and livestock activities. In the past two decades, reforestation with P. patula has led to the establishment of a pine forest plantation, resulting in the isolation of a forest relict of P. pseudostrobus var. coatepecensis, which represents the only remaining stand within the forest plantation matrix (Fig. 1).
Fig. 1 - (a) Blue circle: Pinus pseudostrobus var. coatepecensis relict. Matrix plantation: P. patula stands. Recent patch: < 3 yrs-old P. pseudostrobus var. coatepecensis. (b): Sampling procedure along a 145 m transect with sampling points: 6 each 20 cm, 4 each 1 m, 4 each 10 m, 4 each 25 m. Each point had three subsamples; soil cores were taken with a soil core sampler (15 cm depth, 5 cm diameter).
Experimental design and sampling method
Sampling was carried out along a 145 m transect, which was established from the area near the plantation to the edge of the relict (Fig. 1). Roots were collected from a total of 18 soil samples, distributed as follows: (a) six samples at 20 cm intervals, (b) four samples at 1 m intervals, (c) four samples at 10 m intervals, and (d) four samples at 25 m intervals, according to the methodology described by Villarreal-Ruiz & Neri-Luna ([46]). Soil samples were extracted as soil cores measuring 15 cm in length and 5 cm in diameter using a soil core sampler. Subsequently, the samples were carefully placed in sealed bags and transported in a cooler to the laboratory. The roots were thoroughly washed with sterile distilled water and then transferred to a sieve series with mesh sizes of 1 mm and 2 mm. Once cleaned, the roots were preserved by storing them in liquid nitrogen in a freezer set at -20 °C until further processing.
DNA extraction and sequencing
DNA extraction was carried out on root samples using DNeasy PowerSoil® Kit (Qiagen, Hilden, Germany). This kit is proposed for environmental samples with high humic acid or sediment content. Extractions were performed following the manufacturer’s instructions. Total DNA concentration was measured with Qubit® 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) in the range of 60 ng μl-1 to 100 ng μl-1 for all samples before metagenomic analysis. PCR amplification was performed targeting the internal transcribed spacer ITS2 region and the conserved regions of 5.8S, and 28S rDNAs of the fungi, by using universal primers ITS3 (5′-GCATCGATGAAGAACGCAGC-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) ([45]). The 16S rRNA genes were amplified targeting the V3 and V4 regions, using the bacterial primers Bakt_341F (CCTACGGGNGGCWGCAG) and Bakt_805R (GACTACHVGGGTATCTAATCC) ([43]). DNA amplification conditions were performed as follows: 94 °C for 5 min; 35 cycles at 94 °C for 50 s, 58 °C for 50 s, 72 °C for 50 s; and a final extension of 72 °C for 10 min. After amplification, each PCR product was confirmed using 1% agarose gel, and they were purified and sequenced by Macrogen Laboratory Seoul, Republic of Korea (⇒ https://dna.macrogen.com/). Sequencing was performed on a MiSeq™ Illumina (Illumina, Inc., San Diego, CA, USA).
Bioinformatics
Sequence processing and classification were performed in R studio ver. 1.3.1093. Primers were removed with “cut adapt” software, and low-quality nucleotides were removed using the DADA2 function to output representative sequences. Sequences were qualified, filtered, and trimmed using the “FilterAndTrim” method, for bacteria and fungi sequences ([13]). We used sequences longer than 50 bp. Paired-ends sequences were merged, removing singletons and de novo chimera sequences using the “removeBimeraDenovo” method to denoise sequences into amplicon sequence variants (ASVs). The taxonomy of the ASV was assigned with the Silva_v132 database ([34]) for bacteria and UNITE dataset ([33]) for fungi. The “phyloseq” package was also used to explore the data, create an object, and generate a matrix of ASV abundances and the taxonomy matrix. The resulting ASV table was grouped at the species level.
Diversity analysis
Hill Diversity Indices were calculated for (α) diversity (Shannon diversity, Simpson diversity, richness, and effective number of species) and sample coverage for species richness using the iNEXT package. To visualize taxa distribution along each distance point, we used heat maps at the phylum level and circus plots at the family level. The dominance patterns in each distance sample point were visualized with rank/abundance curves. Diversity (β) was calculated and separated into two components: species replacement (turnover) and species nestedness ([7]). Results were based on pairwise comparisons of each sample point, calculated as Sørensen’s Dissimilarity Index (βsor) and, a dissimilarity analysis (ANOSIM) was performed to analyze the similarity of microbial communities in distance groups (d02 = 0.20 cm, d1 = 1 m, d10 = 10 m, d25 = 25 m). R values close to 1 indicate high dissimilarity. We used indicator species analysis to assess whether ASVs occur in different sample distances.
Soil analysis
In the same way as the root sampling, 18 soil samples were taken along the 145 m transect, as follows: (a) six each 20 cm, (b) four each 1 m, (c) four each 10 m, (d) four each 25 m. We manually removed stones and litter before sampling. Each soil sample was stored, dried at room temperature, and used for analyzing soil organic matter (SOM) and organic C content by the oxidation method; pH was measured in a suspension of soil: deionized water (1: 2 w/v); total N, by micro-Kjeldahl method; the C: N ratio as the index determined by the organic C and total N content; ammonium (NH4+-N) quantification was carried out using Nessler’s reagent ([11]); Nitrates (NO3-N) were measured by the Cataldo’s method ([14]). Exchangeable acidity (Al+3+H+), exchangeable aluminum (Al+3), and total hydrogen (H+) according to Bremner & Mulvaney ([11]); Bray-II determined available P (PO4-3) in soil according to Bray & Kurtz ([10]), and Fe+ was quantified by the digestion of concentrated HCl ([15]).
Statistical analysis
To infer the most influential variables affecting the composition of bacterial and fungal communities, we employed Non-metric MultiDimensional Scaling (NMDS) analysis in the R “vegan” package with Bray Curtis dissimilarity, using the “envfit” function which calculates multiple regression of environmental variables. Here, we used soil variables as dependent and selected ordination axes as an explanatory variable. Significance was tested by permutation test (p = 0.05). All statistical analyses were performed in R ([35]).
Results
Fungal community composition
A total of 2.1 million paired-end raw reads were obtained for 17 samples. One fungi sample (point 2) was removed due to insufficient DNA. After applying quality filters, removing chimeras, and merging paired-end reads, we obtained 908.991 sequences, with an average of 49.471 sequences per sample. The DADA2 pipeline inferred 1.233 fungal ASVs. After filtering out rare ASVs, we identified 995 fungal ASVs, including 212 ASVs belonging to Basidiomycota, 326 ASVs belonging to Ascomycota, and 20 ASVs belonging to Glomeromycota. Among the fungal sequences, 69 ASVs were attributed to ectomycorrhizal (EcM) fungi. The EcM families with the highest relative abundance were Thelephoraceae, Clavulinaceae, Suillaceae, Russulaceae, Elaphomycetaceae, and Amanitaceae (Fig. S1 in Supplementary material).
Along the transect, we observed different functional fungal groups ranging from mutualistic to pathogenic (Fig. 2a). The dissimilarity of the total fungal community between samples at different distances was significant (p = 0.0397) with an R2 = 0.1984. Russula dissimulans Shaffer was associated with samples collected at short (20 cm) distances (p = 0.0275). Tomentellopsis zygodesmoides (Ellis) Hjortstam (p = 0.0165) and Lactarius chrysorrheus Fr. (p = 0.0188) were associated with samples taken at 1 m. Hyaloscypha variabilis (Hambl. & Sigler) Vohník, Fehrer & Réblová (p = 0.0263) and Gymnopilus penetrans (Fr.) Murrill (p = 0.0263) were associated with samples taken at 10 m intervals, while Xenasmatella sp. (p = 0.0164) and Venturia sp. (p = 0.0085), a phytopathogenic genus, were both associated with samples spaced 25 m apart and located close to the remnant edge. Some phytopathogenic fungi, such as Armillaria and Lecanicillium, were found at the beginning of the transect (in the area close to the plantation matrix), but their association with these samples was not significant.
Fig. 2 - Taxonomic composition of fungal communities in root samples of Pinus pseudostrobus var. coatepecensis. (a) Percentages indicate the relative values of paired reads at the genus level. Taxa are grouped based on the ecological function of the fungal taxa. (b) Relative values of the most abundant Amplicon Variant Sequences (ASV). The relative values <1 appear as 0; more than 80% of the relative values <1 were singleton representatives (data not shown).
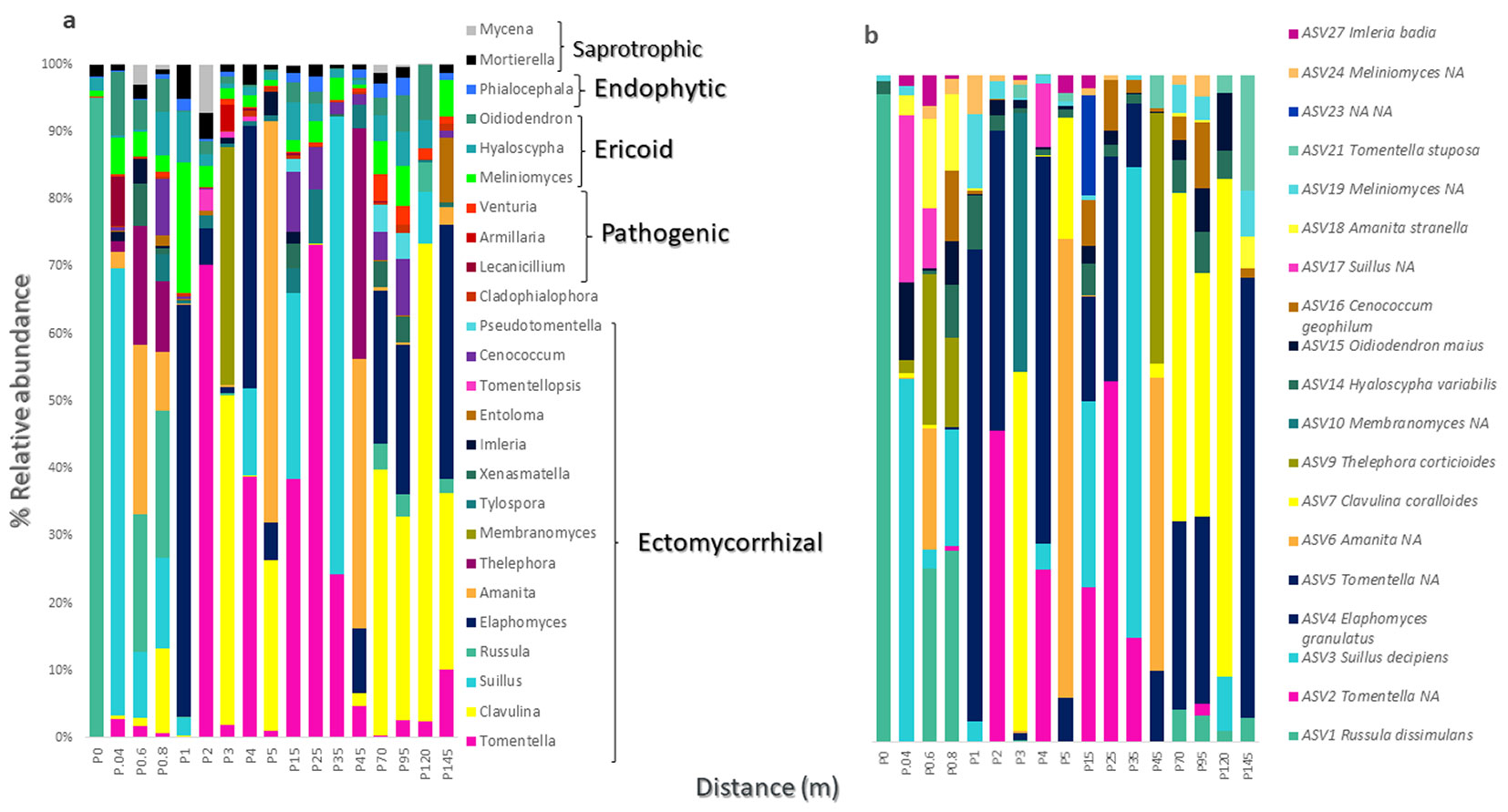
AVS abundance values show that the most abundant fungi were ectomycorrhizal (Fig. 2b), such as R. dissimulans (ASV1), with the highest relative abundance along the transect, 90.13% at the first sampling point. It was followed by Tomentella sp.1 (ASV2), Suillus decipiens (Peck) Kuntze (ASV3), Elaphomyces granulatus Fr., (ASV4), Tomentella sp2 (ASV3), and Amanita sp.1 (ASV5). Ericoid fungi and root endophytes were also observed along the entire transect with low relative abundance percentages.
The abundance curves of the fungal group exhibited a steep gradient at all sampling points, reflecting richness and evenness. The curves of short-distance samples showed similar slopes (Fig. S2 in Supplementary material). Specifically, the 20 cm sample had few species and low species evenness. The samples taken at a 1 m distance from each other displayed similar slopes and contained a greater number of fungal species, as observed at the 2 m and 4 m distance sample points. The range of ASVs at 5 m intervals was lower. The curves generated from the innermost zone of the remnant exhibited a long and shallow slope, indicating high uniformity. Near the edge of the remnant, with a greater distance between samples, a similar slope was observed at the beginning of the transect, with low evenness in all samples.
Fungi, (α) and (β) diversity
The study site had an effective richness (q=0) of 1233 fungal ASV. Diversity was determined with the Shannon index (q=1) with 53 virtual taxa and dominance (q=2) with 23 taxa; the sampling coverage was close to 100%, so it can be inferred that the sampling was sufficiently exhaustive for both fungi and bacteria (Tab. 1). Regarding β-diversity (βsor), species turnover (β-3) was the major component in dissimilarity among fungal assemblages (Fig. S3 in Supplementary material). Species turnover achieved high values, above 0.8 at sample points closer to the edge, and a lower turnover in the innermost part of the remnant. Overall, the sampling points along transects showed a low proportion of shared species yielding high dissimilarity values. The resulting nesting values (βnes) were shallow in all comparisons.
Tab. 1 - Estimation of (α) diversity using Hill numbers, evaluation of microbial representativeness and sample coverage.
| Estimate | Fungal community | Bacterial community |
|---|---|---|
| Effective richness (q=0) | 1.233 | 10.237 |
| Shannon diversity (q=1) | 53 | 6.099 |
| Simpson diversity (q=2) | 23 | 3.759 |
| Sample coverage | 99.9 % | 99.8 % |
Bacterial community composition
A total of 2.2 million bacterial sequences were obtained from 18 samples. After filtering and removing chimeras, 876.010 sequences remained, resulting in a total of 16.233 ASVs. After filtering out rare ASVs, the number was reduced to 10.237. Taxonomically, these sequences were classified into different groups, including 2.996 Acidobacteria ASVs, 3.787 Proteobacteria, 1.306 Verrucomicrobia ASVs, 1.007 Actinobacteria ASVs, and 1.141 Bacteroidetes ASVs (Fig. S4 in Supplementary material). No significant differences were observed in bacterial community dissimilarity between sample distances (p = 0.1205), indicating a low dissimilarity among the communities (R2 = 0.1086). Twenty-six bacterial ASVs were found to be statistically significant in the 20 cm distance group, including Acidothermus sp., Bryobacter sp., Burkholderia-Paraburkholderia sp., Rhodospirillales spp., and Xanthomonadales spp. (p = 0.009). Acidibacter sp. (p = 0.0297) and Granulicella sp. (p = 0.0396) were also among the significant ASVs. In the 1 m distance group, 21 ASVs were associated with the samples, such as Acidobacteria sp., Acidobacterium sp., Solirubrobacterales spp. (p = 0.009), Rhizomicrobium sp. (p = 0.0297), and Bryobacter sp. (p = 0.0495). For samples spaced 10 m apart, 71 ASVs were found, with statistically significant ASVs including Xanthomonadales spp., Rhodospirillales spp., Rhodomicrobium sp., Chitinophagaceae spp., Dyella sp., Acidothermus sp., Verrucomicrobia sp., and Candidatus_methylacidiphilum sp. (p = 0.009). Some bacteria, such as Nevkia (p = 0.0297), Sorangium (p = 0.0396), and Gemmatirosa (p = 0.0297), were exclusively present in this group. In the samples spaced 25 m apart, 18 bacterial ASVs were statistically associated, including Rhizomicrobium sp., Verrucomicrobia sp., and Granulicella spp. (p = 0.009), Bryobacter sp. (p = 0.019), Acidobacteria sp. (p = 0.0297), Acidothermus spp. (p = 0.0495), and Rudaea sp. (p = 0.0495). At the family level, Acidobacteriaceae (18.6%), Solibacteriaceae (7.4%), and Burkholderiaceae (4.3%) were the most abundant along the transect, representing the major components of the bacterial community associated with the roots of P. pseudostrobus var. coatepecensis. Burkholderia, Bryobacter, Acidobacterium, and Acidothermus (Fig. 3a) were the dominant genera along the transect. As expected, numerous ASVs exhibited low abundance, while a few ASVs constituted most of the community (Fig. 3b), such as Bradyrizobium sp.1 (ASV1) and Acidobacteriaceae sp.1 (ASV2).
Fig. 3 - Taxonomic composition of bacterial communities in root samples of Pinus pseudostrobus var. coatepecensis. (a) Percentages indicate the relative values of paired reads at the genus level. (b) Relative values of the most abundant Amplicon Sequences Variant (ASV). The relative values <1 appear as 0; more than 80% of the relative values <1 were singleton representatives (data not shown).
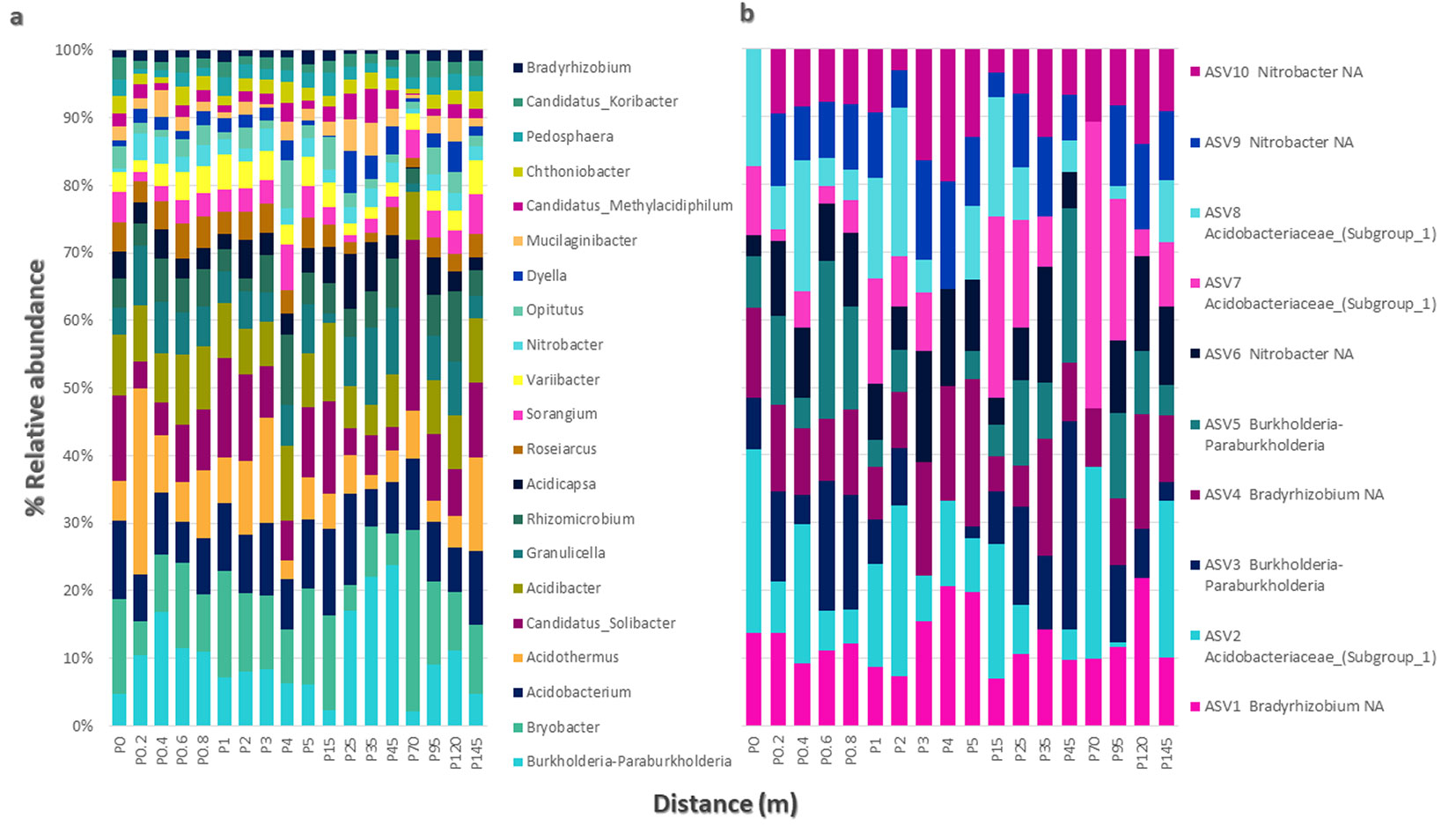
The range-abundance curves showed that the first six sampling points (spaced every 20 cm) displayed similar steep slopes, indicating a higher number of ASVs compared to more distant sampling points and resulting in high evenness (Fig. S5 in Supplementary material). In the inner zone of the remnant, the slopes decreased and aligned with the graph, suggesting community uniformity and lower dominance.
Bacteria, (α) and (β) diversity
The effective richness (q=0) of bacterial ASV was determined to be 10.237, with a diversity (q=1) of 6.099 virtual taxa and a dominance (q=2) of 3.799 bacterial taxa (Tab. 1). In terms of bacterial community dissimilarity (βsor), turnover was found to be the major contributing factor to dissimilarities among bacterial assemblages (Fig. S6 in Supplementary material). The lowest turnover value was observed at the starting point of the transect, but no clear trend in turnover with respect to distance between points was observed. The nesting values (βnes) generally indicated low dissimilarity between samples.
Soil drivers as predictors for microbial community
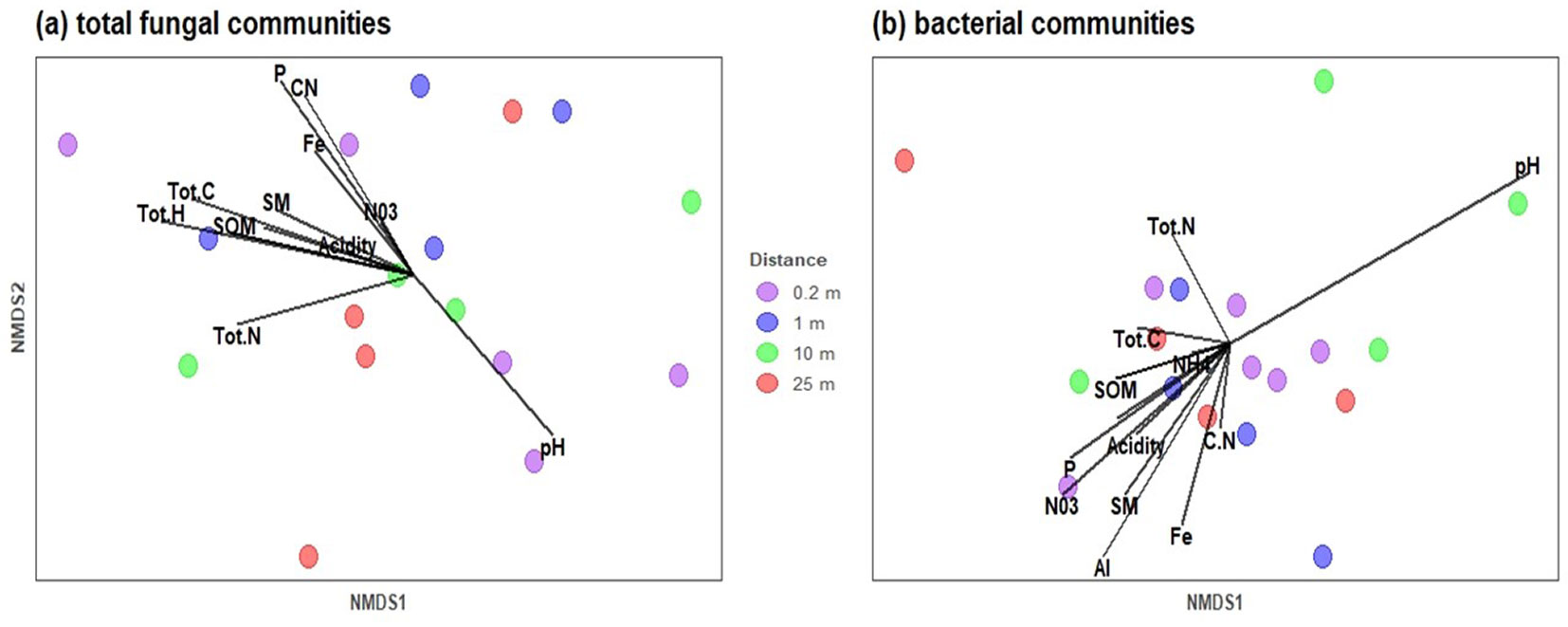
Among the soil drivers, available P (R2 = 0.4469, p = 0.020), total C (R2 = 0.3671, p = 0.033), total H (R2 = 0.4578, p = 0.012), and C:N ratio (R2 = 0.3692, p = 0.033) emerged as the strongest predictors of fungal community composition. In contrast, for the EcM fungal community, no clear correlation between species and soil properties was observed, and no significant predictors were identified. Soil pH (R2 = 0.6051, p = 0.001) was found to strongly influence the composition of the soil bacterial community (Fig. 4).
Fig. 4 - Non-metric multidimensional scaling (NMDS) of (a) fungal communities and (b) bacterial communities at different soil sample distances. The arrow vectors represent the effect of each physico-chemical characteristic of the soil on microbial communities calculated with the “envfit” function. SM: Soil moisture, Acidity, SOM: Soil organic matter, Org C: Organic C, CN ratio, Tot N: Total N, Tot C: Total C, Tot H: Total H, Al+3, Fe+, NO3-: nitrate, NH4+: ammonium, P available, and pH. (ANOSIM, R = 0.1984; p-value = 0.0397).
Discussion
Fungal community diversity and composition
The results of this study demonstrate that the forest relict serves as a reservoir of rich fungal diversity. The EcM fungal community exhibited a higher relative abundance, consistent with previous findings reported by Gavito et al. ([22]). Their study conducted in small pine forest fragments within an agricultural matrix revealed the presence of 60 to 109 EcM OTUs in patches ranging from 100 to 400 m2. However, richness estimation should be carried out carefully, as the analysis method influences species richness and diversity.
The dominant fungal families identified in our study were Thelephoraceae, Clavulinaceae, Suillaceae, and Russulaceae. These findings are consistent with previous studies carried out in Neotropical conifer forests. Argüelles-Moyao et al. ([4]) reported similar dominance patterns in Abies-Pinus forests, where Russulaceae and Clavulinaceae, along with Inocybaceae and Atheliaceae, were dominant families. However, our results differ from those reported in Pinus montezumae Lamb., a species distributed in the same geographic area as P. pseudostrobus, where Atheliaceae, Cortinariaceae, and Sebacinaceae were found to be the most dominant ([38]). Notably, Atheliaceae has been identified as the primary family associated with P. hartwegii Lindl. in Neotropical alpine areas ([6]). Interestingly, in our study, Atheliaceae was not dominant and was represented by only two genera: Tylospora and Taeniospora (anamorph).
Furthermore, our results highlight Thelephoraceae as the most dominant family, comprising four genera: Pseudotomentella, Thelephora, Tomentellopsis, and Tomentella. Among these, Tomentella exhibited the highest species diversity, including Tomentella stuposa (Link) Stalpers, Tomentella radiosa (P. Karst.) Rick, Tomentella coerulea Höhn. & Litsch., and two unidentified species. The Tomentella/Thelephora lineage is widely recognized as one of the most dominant in EcM communities worldwide, also in Neotropical ecosystems ([3]).
It is important to consider the representation of the pathogenic fungal community in our study. We found that Armillaria gallica, a saprobic and facultative pathogenic species known to attack conifer roots, was present at the study site but in low abundance. Importantly, there was no evidence of damage to the trees caused by this pathogen. Previous studies, such as Viswanathan et al. ([48]) have suggested that plant-fungal pathogen interactions tend to decrease in fragmented and smaller patches. In our results, we observed that the proportion of pathogens relative to the total fungal community was 21%, and for ectomycorrhizal (EcM) fungi specifically, it was 12%. This highlights the need for further research on the potential of these forest fragments to be integrated into forestry management plans to reduce and control potential diseases in adjacent plantations.
Regarding β-diversity, the fungal species composition showed a high degree of dissimilarity even at short distances, such as 20 cm. This pattern can be explained by the different dispersal abilities of fungal species, which are influenced more by the forest matrix than by the distance between sampling points. Existing evidence suggests that the dispersal ability of fungal propagules plays a significant role in fungal community turnover. For instance, we observed a higher abundance of Russula species at sampling points closer to the forest matrix. These findings are consistent with the study by Boeraeve et al. ([9]), which reported a higher abundance of Russula species in patches near natural forests, with a decrease in isolated patches. This pattern may be attributed to the contact exploration capacity of Russula species and the limited dispersal ability of some ectomycorrhizal fungi ([41]). Consequently, patches exhibiting a micro-environmental gradient between the forest edge and the interior of the main forest can show high turnover values in fungal communities.
The analysis of relative abundance revealed the dominance of species with short to medium-distance exploration types, including Russula, Amanita, Thelephora, Tomentella, and Elaphomyces. This observation is in line with expectations considering the disturbance background of the study site. According to Correia et al. ([17]), species with such exploration types are more resilient to disturbances because they can quickly regenerate their extraradical hyphal systems.
Bacterial community diversity and composition
The bacterial communities in acidic soils of coniferous forests are often characterized by the dominance of common bacterial phyla, including Acidobacteria, Actinobacteria, Bacteroidetes, Verrucomicrobia, and Proteobacteria ([30]). These same phyla were found to dominate the roots of P. pseudostrobus in our study. Notably, we observed the presence of Acidobacterium, Acidothermus, Burkholderia, and Bryobacter genera in all samples. This study represents the first report on the bacterial diversity associated with P. pseudostrobus. However, it is worth mentioning that certain bacterial genera such as Cohnella, Cupriavidus, Pseudomonas, Stenotrophomonas, and Rhodococcus have been reported as growth promoters in P. pseudostrobus seedlings, based on their isolation from the roots of Abies religiosa, P. hartwegii, and P. montezumae ([23]). It is worth noting that, in our study, the Pseudomonas genus was found to be present in low abundance in the 10 m distance group and did not show any statistically significant association.
To date, there have been limited reports on bacteria associated with Neotropical pines, their roots, and soils. For example, Rivera et al. ([36]) reported that the dominant bacterial phyla in P. patula forests along a land-use gradient were Proteobacteria (40.13%), Actinobacteria (20.15%), and Acidobacteria (14.50%), which aligns with our findings. However, the bacterial community at the genus level differed from those associated with P. pseudostrobus. The aforementioned authors reported Halomonas, DA101, Bacillus, Streptomyces, Rhodoplanes, and Candidatus-Solibacter as the most abundant bacteria, with their abundance being influenced by land use, decreasing in arable soil and increasing in forest soil, irrespective of forest management. Another study identified 498 bacterial isolates from P. chiapensis rhizosphere, representing five genera: Bacillus, Paraburkholderia, Dyella, Luteimonas, and Enterobacter ([18]). Some of these genera, such as Paraburkholderia and Dyella, were also found in our study.
Moreover, the dominant bacteria observed in P. pseudostrobus roots are known to be associated with host-pathogen resistance, such as Acidothermus, which exhibited high abundance along the transect. Zhang et al. ([51]) recently reported a higher abundance of Acidothermus in the roots of three Pinus species (P. taeda, P. caribaea, and P. elliottii), which are known to be resistant to the nematode Bursaphelenchus xylophilus, in comparison to non-healthy pines such as P. massoniana. Another significant bacterial genus found in our study was Burkholderia-Paraburkholderia. These bacterial communities are predominant in Russula spp. sporocarps and act as mycorrhizal helper bacteria, promoting mycorrhizal colonization and hyphal growth ([49]). Our findings are consistent with this, as Burkholderia-Paraburkholderia was associated with short-distance samples (20 cm apart) where Russula dissimulans exhibited higher abundance. Further investigations are necessary to understand the specificity of Burkholderia-Paraburkholderia interactions with Russula species and to elucidate the high biotechnological potential of these bacteria in the development of forest inoculants.
Additionally, Burkholderia species play multiple roles in soil ecosystems and mycorrhizal colonization. They are the principal associates of ectomycorrhizal (EcM) root tips in P. muricata ([31]). Furthermore, a large portion of the bacteria associated with pine root tips belonged to the phylum Acidobacteria, specifically Acidobacterium and Bryobacter genera. The ecological roles of Acidobacterium species are still not well understood, as previously suggested by Kataoka et al. ([26]). These authors identified Acidobacterium sp. in the mycorrhizosphere of Tricholoma matsutake in a P. densiflora forest, but their specific functions remain unknown. Recent evidence suggests that some Acidobacteria genera may be oligotrophic bacteria with slow growth rates, and they may play a key role in nutrient cycling in nutrient-poor soils ([27]). Our results indicate that the bacterial communities associated with pine root tips were not significantly affected by the distance between samples. However, we observed a tendency for certain genera to be associated with specific fungal species along the transect. Further investigations, including fungal-bacterial isolation experiments, are necessary to gain a better understanding of fungal-bacterial symbiosis.
Effects of soil properties on fungal and bacteria communities
We examined the influence of edaphic factors on the composition of fungal and bacterial communities. Our findings revealed that the C:N ratio, total C, H+, and P were the key factors that determined the differences in total fungal community composition. Specifically, the C:N ratio played a significant role in shaping the total fungal communities at short-distance sampling points. Ectomycorrhizal (EcM) fungi dominated the fungal communities, which can contribute to increased N supply through ectomycorrhizal associations, resulting in higher soil C:N ratios ([21]). This preference for N acquisition by EcM fungi over saprophytic species may help predict N-mineralization rates in the soil. Our results align with previous studies that reported a significant correlation between fungal community composition and the C:N ratio, indicating C limitation relative to N ([5]). This could explain the higher abundance of EcM fungal species compared to saprophytic fungi ([24]). The most abundant species along the transect and in short-distance samples was R. dissimulans. Previous studies have also identified this genus as dominant in forest soils, characterized by hydrophilicity and different adaptations in N acquisition ([16]). EcM fungi, such as R. dissimulans, are involved in carbon cycling and transfer among plants, as they possess facultative saprotrophic abilities and rely on their host plants for carbon sources ([19]). NMDS analysis further revealed that total C influenced the entire fungal community composition. In our study, R. dissimulans, T. zygodesmoides, and L. chrysorrheus were statistically associated with samples at short distances. These EcM species, along with the abundant genera Tomentella, Elaphomyces, and Amanita, exhibit a short to medium-distance exploration type. This observation supports the findings of Rog et al. ([40]), who proposed that EcM fungal species with contact exploration types dominate below-ground carbon networks. According to Santini et al. ([42]), P. pseudostrobus forests have a high potential for carbon storage, with soil carbon reservoirs ranging from 42 to 145 Mg SOC ha-1. Further analyses are needed to understand the environmental services provided by ectomycorrhizal networks in these forest relicts within forest plantations.
Additionally, soil phosphorus concentrations (P Bray) influenced fungal communities, particularly EcM fungi ([50]). Different fungal species exhibit varying production of exoenzymes involved in phosphorus hydrolysis. For instance, Russula species exhibit high expression of acid phosphomonoesterases and phosphodiesterases, leading to increased phosphorus availability. As expected, we found that soil pH levels in the range of 4.05 to 4.80 strongly influenced soil bacterial composition. This finding is consistent with the study by Lammel et al. ([29]), which demonstrated a direct correlation between low pH and the abundances of Acidibacter, Acidothermus, and Bradyrhizobium. Ni et al. ([32]) also reported a negative correlation between phyla Proteobacteria and Acidobacteria with soil pH and exchangeable Ca2+, while Actinobacteria, Planctomycetes, Chloroflexi, Nitrospirae, and Gemmatimonadetes showed a positive correlation. Our results suggest that fungal and bacterial species may benefit from resources obtained from forest plantations adjacent to forest relicts. Therefore, considering forest relicts as reservoirs of native microbial diversity in forestry systems could represent a potential alternative to enhance productivity and minimize diseases in forest plantations. Further investigations are needed to analyze the functional role of these natural relicts.
Conclusions
Neotropical forests are severely degraded, and the potential of forest relicts as reservoirs of microbial diversity needs to be further explored and integrated into forest management and production programs. In this study, we identified a total of 212 ASVs (Basidiomycota), 326 ASVs (Ascomycota), 20 ASVs (Glomeromycota), 2.996 ASVs (Acidobacteria), 3.787 ASVs (Proteobacteria), 1.306 ASVs (Verrucomicrobia), 1.007 ASVs (Actinobacteria), and 1.141 ASVs (Bacteroidetes) associated with P. pseudostrobus var. coatepecensis. The EcM fungal communities were dominated by Amanita, Clavulina, Elaphomyces, Russula, and Tomentella genera, while the bacterial communities were dominated by Burkholderia-Paraburkholderia and Bryobacter genera. Our findings demonstrated the sensitivity of microbial communities to edaphic factors, particularly the C:N ratio, total C, available P, and total H+. The β-diversity at small scales was strongly influenced by changes in soil properties and the dispersal ability of microorganisms across the adjacent matrix. The composition and structure of microbial communities can serve as early indicators of ecosystem health and resilience at local scales. Therefore, future research should evaluate the impact of native forest relicts on the conservation of microbial diversity within productive landscapes.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This article is part of the requirements for YB-G to pursue a Doctoral degree by the Doctorado en Ciencias Biológicas y de la Salud from the Universidad Autónoma Metropolitana (UAM), Mexico City. YB-G gratefully acknowledges the financial support from CONACYT (753106). We thank the DNA Sequencing Center-MACROGENE (South Korea) for the metagenomic service. Special thanks to the Laboratorio de Organismos Benéficos de la Universidad Veracruzana for providing infrastructure, materials, and field support. We extend our gratitude to the people who facilitated the fieldwork in the forest plantation, and we are also grateful to the anonymous reviewers for their valuable comments and suggestions.
References
CrossRef | Gscholar
CrossRef | Gscholar
Gscholar
CrossRef | Gscholar
Gscholar
CrossRef | Gscholar
Gscholar
CrossRef | Gscholar
CrossRef | Gscholar
CrossRef | Gscholar
CrossRef | Gscholar
Supplementary Material
Authors’ Info
Authors’ Affiliation
Doctorado en Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana, 09310, Mexico City (Mexico)
Noé Manuel Montaño 0000-0001-5836-9837
Departamento de Biología, División de Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana, Unidad Iztapalapa, Mexico City, 09310 (Mexico)
Facultad de Ciencias Agrícolas, Universidad Veracruzana, Circuito Gonzalo Aguirre Beltrán s/n, Zona Universitaria, 91090, Xalapa, Veracruz (Mexico)
Corresponding author
Paper Info
Citation
Baeza-Guzmán Y, Camargo-Ricalde SL, Trejo Aguilar D, Montaño NM (2023). Fungal and bacterial communities in a forest relict of Pinus pseudostrobus var. coatepecensis. iForest 16: 299-306. - doi: 10.3832/ifor4284-016
Academic Editor
Alberto Santini
Paper history
Received: Dec 07, 2022
Accepted: Sep 13, 2023
First online: Nov 09, 2023
Publication Date: Dec 31, 2023
Publication Time: 1.90 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2023
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 19433
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 14238
Abstract Page Views: 2916
PDF Downloads: 1828
Citation/Reference Downloads: 1
XML Downloads: 450
Web Metrics
Days since publication: 999
Overall contacts: 19433
Avg. contacts per week: 136.17
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Jul 2026)
Total number of cites (since 2023): 4
Average cites per year: 1.00
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Ectomycorrhizal fungal community in mature white poplar plantation
vol. 14, pp. 540-547 (online: 26 November 2021)
Research Articles
Ectomycorrhizal diversity in a mature pedunculate oak stand near Morović, Serbia
vol. 16, pp. 345-351 (online: 22 November 2023)
Review Papers
Soil fungal communities across land use types
vol. 13, pp. 548-558 (online: 23 November 2020)
Research Articles
Root colonization and growth response of Pinus nigra seedlings to three types of ectomycorrhizal inoculum
vol. 19, pp. 269-275 (online: 23 July 2026)
Research Articles
Potential spread of forest soil-borne fungi through earthworm consumption and casting
vol. 8, pp. 295-301 (online: 26 August 2014)
Research Articles
Soil fauna communities and microbial activities response to litter and soil properties under degraded and restored forests of Hyrcania
vol. 14, pp. 490-498 (online: 11 November 2021)
Research Articles
Effect of plant species on P cycle-related microorganisms associated with litter decomposition and P soil availability: implications for agroforestry management
vol. 9, pp. 294-302 (online: 05 October 2015)
Research Articles
Mapping fungi from below ground: online genetic resources and ectomycorrhizal geographic distributions
vol. 4, pp. 252-255 (online: 13 December 2011)
Research Articles
Ectomycorrhizal fungal community associated with autochthonous white poplar from Serbia
vol. 9, pp. 330-336 (online: 12 November 2015)
Short Communications
Not all long-distance-exploration types of ectomycorrhizae are the same: differential accumulation of nitrogen and carbon in Scleroderma and Xerocomus in response to variations in soil fertility
vol. 14, pp. 48-52 (online: 18 January 2021)
iForest Database Search
Search By Author
Search By Keyword
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords
PubMed Search
Search By Author
Search By Keyword







