
Real-Time PCR for Ceratocystis platani detection: in-depth validation to assess the diagnostic potential and include additional technical options
iForest - Biogeosciences and Forestry, Volume 11, Issue 4, Pages 499-509 (2018)
doi: https://doi.org/10.3832/ifor2527-011
Published: Jul 18, 2018 - Copyright © 2018 SISEF
Research Articles
Abstract
A high-performing detection method is essential to safeguard those countries that are still unaffected by canker stain, a devastating disease of Platanus spp. caused by Ceratocystis platani. We previously developed EvaGreen and Taqman-based Real-Time PCR to detect this pathogen, but in-depth validation is needed to guarantee users about its effectiveness and promote its utilization. In this work we present a validation study designed according to EPPO standards, focusing on the analytical and diagnostic sensitivity and specificity. We extend its technical application using SYBR Green. By performing standard curves and eight-replication-based experiments, we established the detection limit at 3 fg C. platani gDNA per PCR reaction. The repeatability and the operator-based reproducibility of the Real-Time PCR was demonstrated. Different gDNA extraction events by different operators and different gDNA extraction modalities did not affect the detection limit. The detection limit threshold cycle was earliest with SYBR Green, followed by Taqman, and EvaGreen. Spiking 6 µl DNA extractions of uninfected, necrotized wood with 3 fg C. platani gDNA confirmed the detection limit: 3 fg C. platani gDNA per PCR reaction, i.e., 0.5 fg gDNA per µl of wood extract. The assays tolerated 6 µl of necrotic C. platani-infected wood extracts without inhibition except for long-dead wood samples, while the 2 µl dose consistently allowed for successful detection. Detection of the pathogen in infected samples showed the highest diagnostic sensitivity with the SYBR Green assay. Agarose gel electrophoresis and staining was validated for visualizing amplicons, even at the detection limit. The specificity of the method was tested against 23 isolates representing the diversity of Ceratocystidaceae, and most species were not detected at 5 ng gDNA. However, some South American strains of the C. fimbriata complex were detected at doses as low as 5 fg. The method remains specific for C. platani detection as no other Ceratocystidaceae are known to colonize plane tree and the species within the geographic range of canker stain of plane tree were only detected at 500 pg or more gDNA. This work paves the way for a performance study of inter-laboratory comparisons.
Keywords
Canker Stain, Real-Time PCR, Validation, EvaGreen, Taqman, SYBR Green
Introduction
Canker stain is by far the most destructive disease of Platanus spp. The disease is caused by the ascomycetous fungus Ceratocystis platani (J. M. Walter) Engelbr. & T. C. Harr., a host-specialized species within the Latin American Clade (LAC) of the C. fimbriata Ell. & Halst. complex, now recognized as the LAC of the genus Ceratocystissensu stricto ([3], [2], [25]). Infection of wounds of the root or stem results in colonization of the sapwood with xylem staining, as in Ceratocystis wilt diseases, but there may also be necrosis of the cambium and living bark tissues (cankers), resulting in the death of the trees ([23], [6]).
C. platani is native to the southeastern USA, but is also found in Modesto, California (USA) and in southern Europe to Asia Minor ([21], [6], [25]). Due to the damage caused in Europe, C. platani was included in the EPPO (European and Mediterranean Plant Protection Organization) A2 list of quarantine organisms ([16], [17], [20]). In Italy, a ministerial decree established mandatory monitoring, prophylaxis and eradication measures to safeguard plane trees ([14]).
A PCR-RFLP method was first developed for identifying isolates of species in the Ceratocystidaceae ([27]). Real-Time PCR methods for C. platani detection developed more recently ([24], [12]). These methods represent the first advanced molecular tools for an effective application of sanitary measures and for studies on epidemiology and the resistance/susceptibility levels of Platanus spp. The method by Luchi et al. ([12]) is based on Taqman minor groove binder (MGB) targeting the cerato-platanin gene and the internal transcribed spacer 2 (ITS2). The ITS2-targeted assay revealed highly sensitive (detection of 2 fg µl-1 DNA extracted from axenic culture = 10 fg/PCR reaction), and it successfully detected C. platani airborne inoculum at 10 conidia ml-1. Thus, the method was specifically designed to monitor and quantify the pathogen at low levels, which is particularly useful for monitoring pathogen levels during sanitation cuttings and investigating the airborne transmission of the pathogen.
The Real-Time PCR assays by Pilotti et al. ([24]) have been included in the EPPO protocol for in planta detection of C. platani ([19]). They are based on EvaGreen dye or a Taqman probe and are targeted to ITS1. These assays proved effective and robust, with high analytical (ASe) and diagnostic sensitivity (DSe), and they were suitable for pathogen quantification in wood. They had an amplification efficiency of nearly 100% when applied both to freshly-infected wood and wood from naturally and artificially infected trees that had been dead for a long time. There was no cross-reaction with a wide range of wood-inhabiting fungi. Regarding the analytical sensitivity, the assay detected 100 ag of the target amplicon-containing plasmid (pDNA) per PCR reaction, corresponding to 20 copies of the target amplicon and 10 fg fungal genomic DNA (gDNA) per PCR reaction ([24], [19]). Overall, these features render this method specifically suited for in-wood detection both for diagnostic purposes and for research on host resistance. Both methods ([24], [12]) have the potential to be applied each for the election use of the other, provided they successfully pass through the required validation processes.
New plant pests are becoming established worldwide, and this has prompted international plant health organizations to fund and coordinate research aimed at developing effective molecular diagnostic methods in order to carry out control measures. In Europe, EPPO and EUPHRESCO (EUropean PHytosanitary RESearch COordination) are promoting this goal by launching studies on diagnostic methods for quarantine and emerging phytosanitary pests. Producing robust validation data has thus become crucial for any method to be included in the official diagnostic EPPO protocols, which are frequently updated as additional validation data become available (⇒ https://www.eppo.int/News&Events/euphresco.htm).
In this paper we present an in-depth validation study designed according to the EPPO guidelines for validation of diagnostic methods ([18], [20]). The focus was on the analytic and diagnostic sensitivities of EvaGreen and Taqman assays. In addition to Eva Green, we validated a low-cost alternative, a SYBR Green assay, and we included a qualitative PCR test with agarose gel electrophoresis as an alternative to Real-Time PCR. Lastly, we determined the diagnostic specificity (DSp) of the assays against representative members of Ceratocystidaceae.
Material and methods
Definitions and objectives
An important issue of this work is the validation of the ASe for pure fungal gDNA and for wood meta DNA (meDNA). We define ASe as the lowest gDNA quantity of the target organism that is detected in the PCR reaction (which we refer to as DQP-Ase: DNA Quantity per PCR reaction - ASe) and in the sample to be tested, expressed per microliter (which we refer to as DQS-Ase: DNA Quantity in the Sample to be tested - ASe).
We introduce the distinction between in-mycelium and in-necrotic-wood, for which ASe refers to gDNA extracted from a pure culture of the microorganism and to meDNA extracted from the necrotic wood, respectively.
Given that these distinctions are not trivial, we provide some explanations. DQS-ASe may vary according to the volume of the tested sample (TS) added to the reaction mixture and the total volume of reaction mixture. The larger the TS volume, the lower the target gDNA quantity per µl of TS (i.e., DQS-ASe). If TS derives from a pure fungal culture, the distinction between DQP-ASe and DQS-ASe is merely conceptual. In principle, the equation in-mycelium DQP-ASe = in-mycelium DQS-ASe × number TS microliters, is valid nevertheless. Note that in this equation in-mycelium DQP-ASe remains constant and in-mycelium DQS-ASe varies in relation to the TS volume. However, it makes sense to exploit DQS-ASe if TS is a DNA extract from a tissue source, i.e., necrotic wood, in which case the template volume may contain natural PCR inhibitors derived from the plant tissue. Thus, if some inhibition takes place, then DQP-Ase is no longer constant and its value increases proportionally to the TS volume (each TS volume has a different value of DQP and DQS-ASe), i.e., the process becomes less sensitive. The presence of the pathogen in the host plant may be quantitatively heterogeneous, leading to variation in fungal DNA titer in various subsamples. However, extracting from large plant samples and adding large volumes of the extract to the Real-Time PCR reaction mixture without encountering inhibitory effects should enhance the capacity of the method to detect the pathogen at low titer.
We had seven key objectives in carrying out this work: (i) to determine the detection limit for in-mycelium DQP-Ase in more detail than we had done previously by inferring standard curves based on narrower dilution windows (5-fold serial dilutions) and to validate it by testing for repeatability and reproducibility, as suggested in ISO/IEC standard 17025 guidelines ([18], [20]); (ii) to determine the in-necrotic-wood DQP and DQS-ASe in order to evaluate if the discoloration in the wood has inhibitory effects; we specifically aimed to assess the capacity of Real-Time PCR assays to tolerate a volume of necrotic wood extract as high as 6 µl vs. the canonical dose of 2 µl; (iii) to compare the use of SYBR Green to EvaGreen and validate it, as specifically requested by some Italian phytosanitary services; (iv) to further validate the diagnostic sensitivity of the Real-Time assays with a focus on naturally-infected, mature trees in comparison with a rapid microscope assay ([23]); (v) to evaluate the tendency of the primers to undergo dimerization and give false positive reactions; (vi) to validate the suitability of agarose gel electrophoresis for amplicon detection in conventional PCR runs, as specifically requested by the Italian phytosanitary services; (vii) to test primers and probe for diagnostic specificity (DSp) within Ceratocystidaceae in order to identify potential problems of cross-reactions with other species or identify the potential to use the system with other taxa.
As specified above the development and validation of some technical options were established in the round table discussions with Italian phytosanitary services, in the frame of the project ASPROPI (Azioni a Supporto della PROtezione delle Piante) financed by the MIPAAF (the Italian Ministry of Agricultural and Forestry Policies).
DNA extraction and Real-Time PCR
Extraction and quantification of DNA from healthy and C. platani-infected plane tree wood has been carried out as previously described ([24], [19]). Genomic DNA was extracted from mycelium of C. platani isolate C.P.32, which was isolated from an infected tree located in Rome. The isolate was grown on potato dextrose agar (PDA, Oxoid Unipath Ltd, Basingstoke, Hampshire, England) or on sterile cellophane discs (BIO-RAD, Hercules, CA, USA) layered on PDA plates and collected by scraping with a sterile pipette tip.
Real-Time PCR amplifications were carried out on a CFX96 Real-Time PCR Detection System equipped with CFX Manager™ software version 3.1 (BIO-RAD). Primers and probe were as previously described ([24], [19]) and were synthesized by Invitrogen and Metabion, respectively. We used EvaGreen and Taqman assays with SsoFast™ EvaGreen® (BIO-RAD - [24]) and SsoAdvanced™ Universal Probes Supermix (BIO-RAD), respectively. The probe was used at a concentration of 0.3 µmol l-1. For the SYBR Green assay, the SsoAdvanced™ Universal SYBR® Green Supermix (BIO-RAD) was used.
Details on DNA extraction, primers and probe sequences, supermix composition and Real-Time PCR thermal cycling, are given in Appendix 1 (Supplementary material). Thermal cycling conditions included an initial denaturation at 96 °C for 3 min, 40 cycles at 95 °C for 10 s (denaturation) and 66 °C for 20 s (annealing/extension); and a final extension at 72 °C for 5 min (for DNA-binding dyes only). Control treatments for Real-Time PCR were reactions with: (i) no-template; (ii) healthy and (iii) C. platani artificially-infected plane tree wood; and (iv) C. platani gDNA isolate C.P. 32.
Testing the risk of primer dimerization
To confirm that the primers were not subject to reciprocal annealing and dimer formation (a possible false positive signal in any dye-based Real-Time PCR), we ran EvaGreen and SYBR Green reactions in a thermal gradient experiment using the following annealing temperatures: 66, 60.5, 56.9 and 51 °C. The reactions contained each primer at 0.5 μmol l-1 and no DNA template. Five replications were tested for each temperature and each dye. The experiment was repeated twice.
Determination and validation of in-mycelium DQP-ASe
To determine the DQP-ASe, we tested eight 5-fold dilution series corresponding to the following C. platani gDNA quantities per PCR reaction: 9.37 pg, 1.87 pg, 375 fg, 75 fg, 15 fg, 3 fg, 0.6 fg, and 120 ag. Each dilution series was assayed in triplicate, and the standard curve experiment was repeated three times with each of the three assays. The detection limit was determined to be the lowest DNA quantity/concentration that was detected consistently at a Ct equal to or lower than the highest acceptable threshold cycle (37.9 - [24]).
To validate the repeatability of DQP-ASe, we performed three experiments for each of the three assays at the limit of detection and at a five-fold higher concentration. Eight replications were used for each assay, concentration and experiment. To validate the reproducibility, the above-described set of experiments was performed by two different operators. We thus performed a total of six experiments for each assay and concentration. The DQP-ASe was also validated by testing the gDNA of five additional C. platani strains, each in a single experiment performed by one operator, at the limit of detection and with eight replications.
An additional set of experiments was performed in order to validate the detection limit in relation to the different modalities of collecting the fungal mycelium, namely, the agar vs. the cellophane modalities. We also investigated whether different extraction events, conducted by two distinct operators, would give a different detection limit. These experiments were conducted with Taqman assay. The operator performed three experiments (with eight replications in each) on each type of extracted gDNA and at 3 and 15 fg gDNA per PCR reaction.
Melting temperature of target amplicon and agarose gel analyses
Dissociation runs were applied downstream of the EvaGreen and SYBR Green assays using amplicons derived from reactions charged with 0.5 ng C. platani gDNA or 1 pg of the PCR target-containing plasmid (pCR®2.1, Invitrogen; see Appendix 1 in Supplementary material for details on cloning - [24]). Dissociation runs were performed with an increase of 0.1 °C, from 55 to 95 °C. Amplifications and dissociations were conducted on 10 replications for each type of template, and the experiment was repeated twice for each dye.
To assess the potential of using the amplification protocol in qualitative PCR (agarose gel electrophoresis), we analysed EvaGreen and SYBR Green Real-Time PCR products on 2% agarose gels. We analysed the amplification products derived from the reactions of a typical standard curve experiment, namely, runs that were charged with DNA ranging from 9.37 pg to 3 fg gDNA (five-fold dilution series). Each gDNA quantity was assayed in triplicate, and each dye-based assay was repeated twice.
We also analysed the amplification products obtained from samples of naturally-infected trees, especially samples that produced late fluorescent signals in Real-Time PCR, thus with a low pathogen titer. For each sample and assay, 2 and 6 µl of meDNA were tested in triplicate. After agarose electrophoresis, gels were stained with GelRed (Biotium, Fremont CA, USA) according to the manufacturer’s instructions. DNA bands were visualized with a Gel DocTM XR+ System (BIO-RAD) before and after GelRed staining.
Determination and validation of in-necrotic-wood DQP and DQS-ASe
The living sapwood of plane trees responds to wounding and colonization by C. platani or other pathogens with a series of host reactions that results in discoloration ([6]), and this necrotic tissue contains a number of compounds that could inhibit PCR. The in-necrotic-wood validation of DQP and DQS-ASe was carried out following two approaches. In the first approach, 2 or 6 µl of extracts from naturally or artificially C. platani-infected samples were added to PCR mixtures. In the second approach, a PCR reaction mixture containing 6 µl C. platani free-necrotic wood extract was spiked with 2 µl containing 3 fg of C. platani gDNA. The C. platani free-necrotic wood extract was a meDNA extract of necrotic sapwood of two plane trees showing cankers and fruiting bodies typical of Fomitiporia mediterranea ([22]). The wood was collected from the canker reaction zone, and fungal isolation and ITS sequencing confirmed the necrotic wood was colonized by F. mediterranea (data not shown). After DNA extraction, the samples were confirmed to be C. platani-free by Real-Time PCR. The extracts were pooled and used as described above. The experiment included eight replications, was repeated three times, and was performed with SYBR Green assay by a single operator.
Validation of DSe and DSp
For validation of DSe, samples were collected by phytosanitary inspectors of the Latium region (central Italy) from 28 trees showing symptoms of canker stain. We assessed a posteriori that each sampled tree showed typical canker stain symptoms, but we processed the samples whether the necrotic wood tissue appeared to be freshly killed or if it appeared to have been dead a long time. Each sample was tested with all three assays by adding 2 or 6 µl of the wood extraction to the PCR reaction (in duplicate/triplicate). Each wood sample was also analyzed with the microscope assay ([23]).
With regard to DSp, 23 isolates representing species and strains of the family Ceratocystidaceae (Tab. 1) were tested with the EvaGreen and Taqman assays in order to evaluate the capacity of primers and probe to differentiate species and strains within this taxonomic group. Template for Real-Time PCR was extracted from isolates grown on malt yeast extract agar (MYEA: 2% malt extract, 0.2% yeast extract, 2% agar) for 4-5 days at room temperature using the Wizard® Genomic DNA Purification Kit (Promega Corporation, Madison, WI, USA). The ethanol pellet was suspended in 50 µl of DNA rehydration solution, and the DNA concentration of the preparation was determined with a Qubit 2.0 Fluorometer. The concentration of each extraction was adjusted with H2O to a working stock of 1 ng µl-1 gDNA.
Tab. 1 - Average Ct values for detection or samples not detected (ND) using two detection systems (EvaGreen/Taqman Probe) at various amounts of genomic DNA extracted from members of the Ceratocystidaceae. Each concentration was tested at least twice. (NT): Concentration not tested; (LAC): Latin American Clade; (AAC): Asian-Australasian Clade; (NAC): North American Clade; (AFC): African Clade of Ceratocystis. Parentheses indicate inconsistent detection at that concentration, and the value given is the average for the positive reactions only. Horizontal lines divide the species in groups that gave no, weak and clear cross-reaction signals.
| Genus and species (Host, Country, Clade of C. fimbriata complex) | Isolate No. | Genomic DNA per 20 µL Reaction | ||||||
|---|---|---|---|---|---|---|---|---|
| 5 ng | 500 pg | 50 pg | 5 pg | 500 fg | 50 fg | 5 fg | ||
| C. fimbriata (Mangifera, Brazil, LAC) |
C994 (CBS 600.70) |
- | - | - | 24.2/23.5 | 28.8/28.1 | 33.4/32.0 | 36.9/36.6 |
| C. cacaofunesta (Theobroma, Brazil, LAC) |
C1983 (CBS 115172) |
- | - | - | 25.3/27.5 | 30.1/30.5 | 34.0/33.9 | (37.0)/37.9 |
| C. fimbriata (Eucalyptus, Brazil, LAC) |
C1442 (CBS 115174) |
- | - | 25.2/24.2 | 28.8/26.6 | 32.9/31.3 | (35.4)/34.7 | ND/ND |
| C. fimbriata (Coffea, Venezuela, LAC) |
C1195 (CBS 146.53) |
- | - | 24.9/22.5 | 29.0/27.5 | 33.7/32.7 | (35.8)/(36.5) | ND/ND |
| C. fimbriata (Ipomoea, USA, LAC) |
C1416 | - | 34.0/33.6 | (35.6)/(35.2) | ND/ND | - | - | - |
| C. colombiana (Coffea, Colombia, LAC) |
C1543 (CBS 135861) |
- | 31.2/NT | (34.1)/37.1 | ND/ND | - | - | - |
| C. ficicola (Ficus, Japan, AAC) |
C1355 (KFCF 9001) |
- | 33.8/36.8 | (37.8)/ND | ND/ND | - | - | - |
| C. variospora (Quercus, USA, NAC) |
C1846 (CBS 114714) |
- | - | 37.3/35.7 | ND/ND | - | - | - |
| C. albifundus (Acacia, South Africa, AFC) |
C1060 | - | 37.0/35.2 | ND/ND | - | - | - | - |
| C. harringtonii (Populus, USA, NAC) |
C89 (CBS 114725) |
35.6/34.8 | ND/36.76 | ND/ND | - | - | - | - |
| Endoconidiophora resinifera | C662 (CBS 100202) |
31.8/34.5 | (34.7)/ND | ND/ND | - | - | - | - |
| E. coerulescens | C693 (CBS 489.80) |
ND/(35.0) | ND/ND | - | - | - | - | - |
| Ambrosiella xylebori | C3051 (CBS 110.61) |
37.6/35.6 | ND/ND | - | - | - | - | - |
| Thielaviopsis radicicola | C869 (CBS 114.47) |
(36.3)/ND | ND/ND | - | - | - | - | - |
| Ceratocystis polychroma (Syzygium, Indonesia, AAC) |
C2240 (CBS 115775) |
ND/ND | - | - | - | - | - | - |
| C. smalleyi (Carya, USA, NAC) |
C684 (CBS 114724) |
ND/ND | - | - | - | - | - | - |
| Chalaropsis populi | C1368 (CBS 484.71) |
ND/ND | - | - | - | - | - | - |
| Chalaropsis thielavioides | C1379 (CBS 180.75) |
ND/ND | - | - | - | - | - | - |
| Endoconidiophora virescens | C252 (CBS 128998) |
ND/ND | - | - | - | - | - | - |
| Thielaviopsis paradoxa | C1003 (CBS 374.83) |
ND/ND | - | - | - | - | - | - |
| Ceratocystis adiposa | C1014 (CBS 147.53) |
ND/ND | - | - | - | - | - | - |
| Huntiella moniliformis | C1007 (CBS 204.90) |
ND/ND | - | - | - | - | - | - |
| Bretziella fagacearum | C517 (CBS 129241) |
ND/ND | - | - | - | - | - | - |
For each species or strain, a 10-fold dilution series at 5 ng to 5 fg per reaction was separately tested with each assay method in order to identify the lowest gDNA concentration that could yield a detectable signal (Ct<38.0). In general, two to three separate experiments were run for each isolate at a range of concentrations that included at least the minimum detected concentration and one dilution greater and one dilution less. For some isolates, three serial dilutions were tested above the detection limit in order to evaluate linearity of the response. In each experiment, the same concentration ranges of C. platani gDNA (extracted from isolate CBS 129000) were included as the positive control.
Statistical analysis
Statistical analyses were performed to assess differences among groups of Ct values representing separate experiments (repeatability), different operators (reproducibility), extraction modalities and extraction events, as well as the different PCR assays. One-way analysis of variance (ANOVA) of completely random design was carried out using the Student-Newman-Keuls test as a multiple testing procedure (post-hoc analysis). When a data group violated ANOVA assumptions, we used Welch’s test coupled with Tukey as post-hoc (normal distribution, heteroscedasticity), Kruskal-Wallis coupled with Mann-Whitney Bonferroni corrected (non-normal distribution, homoscedasticity), or Friedman coupled with Wilcoxon (non-normal distribution, heteroscedasticity). The results of pairwise comparisons were evaluated at the 99% confidence level. See also Tab. S1 in Supplementary material for more details. All Ct group data used in the statistical analyses are reported in Tab. S2 (Supplementary material). STATGRAPHICS® Centurion XVI and PAST version 2.17 were used for the analyses.
Results
Tendency of the primers to dimerize, and characterization of in-mycelium DQP-ASe
Amplifying no-template reactions at different temperatures with EvaGreen or SYBR Green did not lead to any amplification product. This was demonstrated by the absence of any fluorescent signals above the threshold line throughout the PCR run and by the dissociation run. Thus, there were no false positives due to dimerization by the primers during the amplification process.
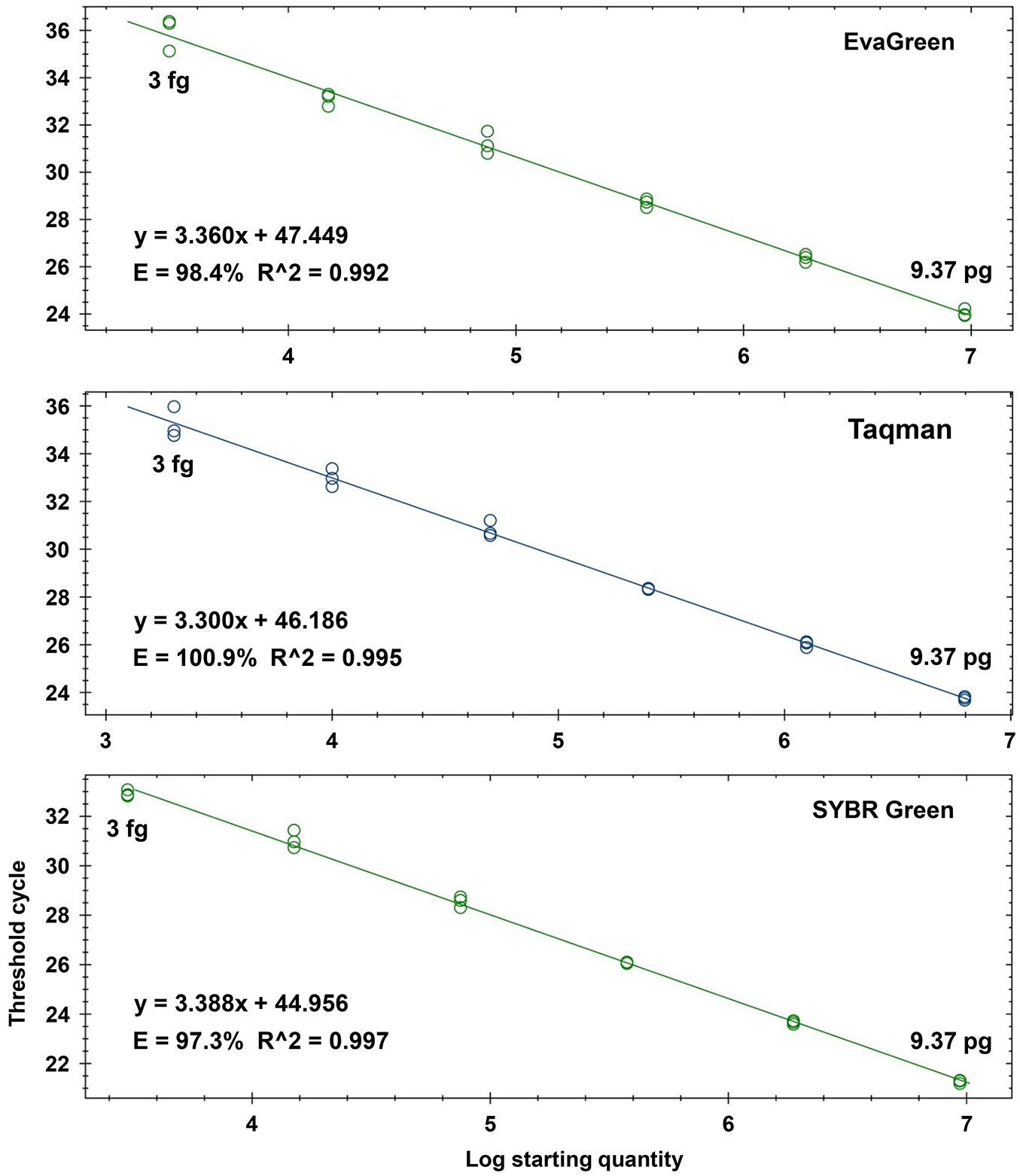
Standard curve experiments showed a large dynamic range for EvaGreen, Taqman and SYBR Green assays. The detection limit with all three assays was 3 fg per PCR reaction, made by adding 2 µl of 1.5 fg µl-1 gDNA to the tested sample (TS - Fig. 1). This result was confirmed three times for each assay. The performance parameters of all the standard curve experiments were in line with a maximum or very high amplification efficiency (E) and repeatability (R2 - Tab. S3 in Supplementary material). Quantities less than 3 fg gDNA were detected, though not in a repeatable manner (data not shown).
Fig. 1 - Standard curves aimed at identifying the detection limit of the Real-Time PCR assays. The curves were obtained through testing eight 5-fold dilution series of Ceratocystis platani gDNA, i.e., from 9.37 pg to 120 ag (including 3 fg) per PCR reaction. We excluded the two lowest dilutions from the standard curve because they were not detected repeatedly or were not detected at all. gDNA was extracted from a pure culture directly grown on PDA.
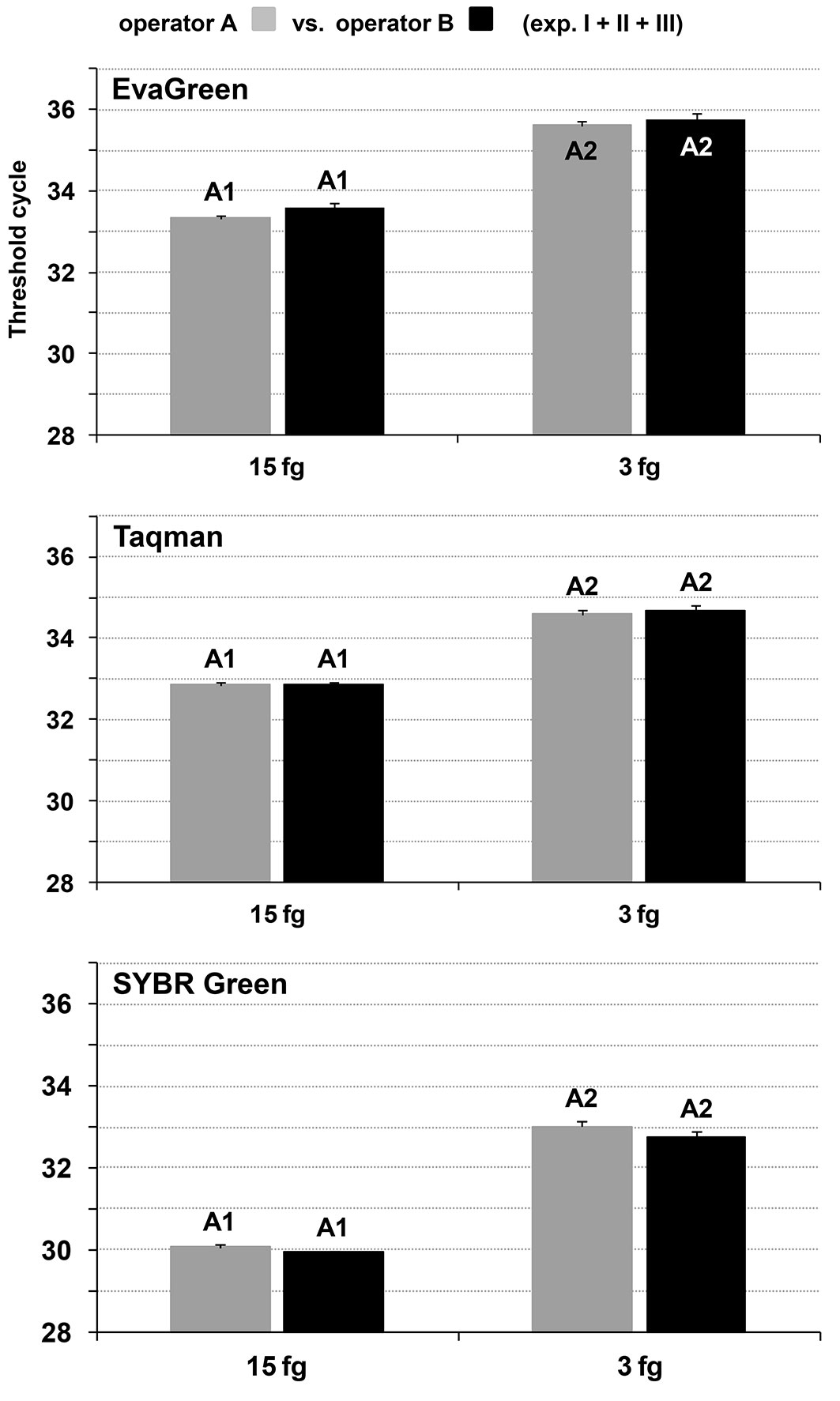
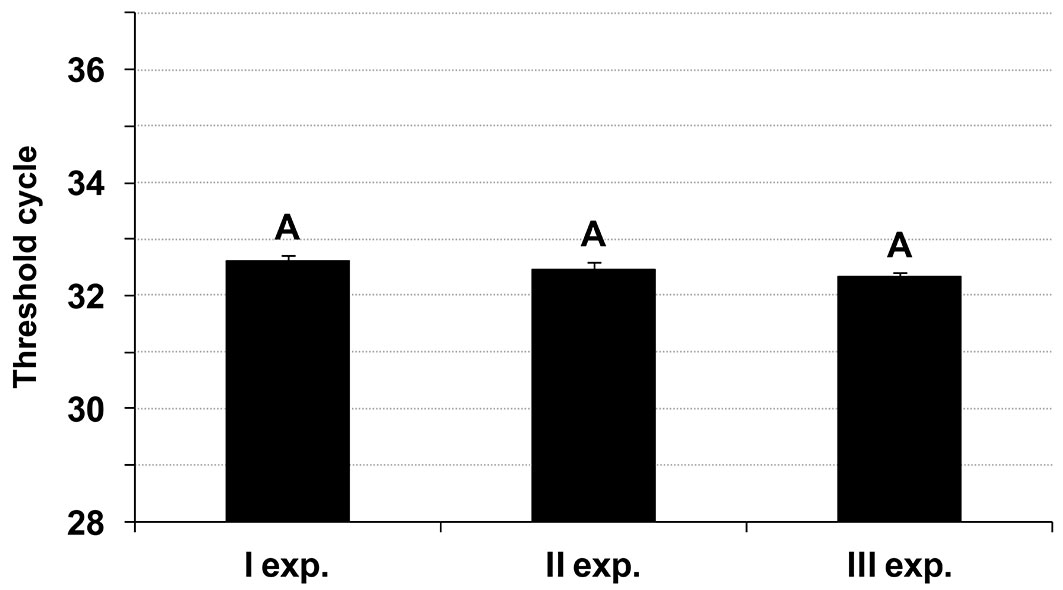
The detection of 15 and 3 fg per PCR reaction (7.5 and 1.5 fg µl-1 TS) in the 8-replication-based experiments by the two operators was “qualitatively” repeatable and reproducible since the expected fluorescent signals were always obtained. They were also confirmed “quantitatively” in that there was no statistically significant difference among the mean Ct values from the various experiments (repeatability, 8 values per dataset) and operators (reproducibility, 24 values per dataset = 3 experiments × 8 replications - Fig. 2; see also Fig. S1, Tab. S1 and Tab. S4a in Supplementary material).
Fig. 2 - Inter-operator reproducibility of the detection limit of the three Real-Time PCR assays. Detection limit refers to the lowest quantity of Ceratocystis platani gDNA detected per PCR reaction (i.e., in-mycelium DQP-ASe). Each bar represents the mean of 24 values (see also Fig. S1 in Supplementary material). Letters indicate homogeneous groups (99.0% confidence level). Numbers indicate the independent statistical analyses. Vertical lines represent the standard error of the mean.
Interestingly, a comparison of the three assays (48 values for each assay = 3 experiments × 8 replications × 2 operators) at both 3 and 15 fg per PCR reactions showed that mean Ct values were significantly different among the three assays, with values from SYBR Green and EvaGreen assays being the lowest and the highest, respectively. This result clearly suggests that the assays are slightly but significantly different in terms of in-mycelium DQP-ASe. The detection of 3 fg gDNA with SYBR Green compared with EvaGreen showed a mean gap of 2.8 Ct values. At 15 fg, the mean gap was 3.5. The differences between SYBR Green and Taqman were 1.7 and 2.9 for 3 and 15 fg, respectively (Fig. 3a, Tab. S4a).
Fig. 3 - Comparison of detection limits (in-mycelium DQP-ASe) of EvaGreen, Taqman and SYBR Green Real-Time PCR. (a) Each bar represents the mean of 48 values derived from combining experiments I, II and III of operators A and B (see also Fig. 2 and Fig. S1). (b) Comparison of the detection limits of the three assays using additional C. platani strains; each bar represents the mean of eight values obtained in a single experiment performed by a single operator. Letters indicate homogeneous groups (99.0% confidence level). Numbers indicate the independent statistical analyses. Vertical lines represent the standard error of the mean.
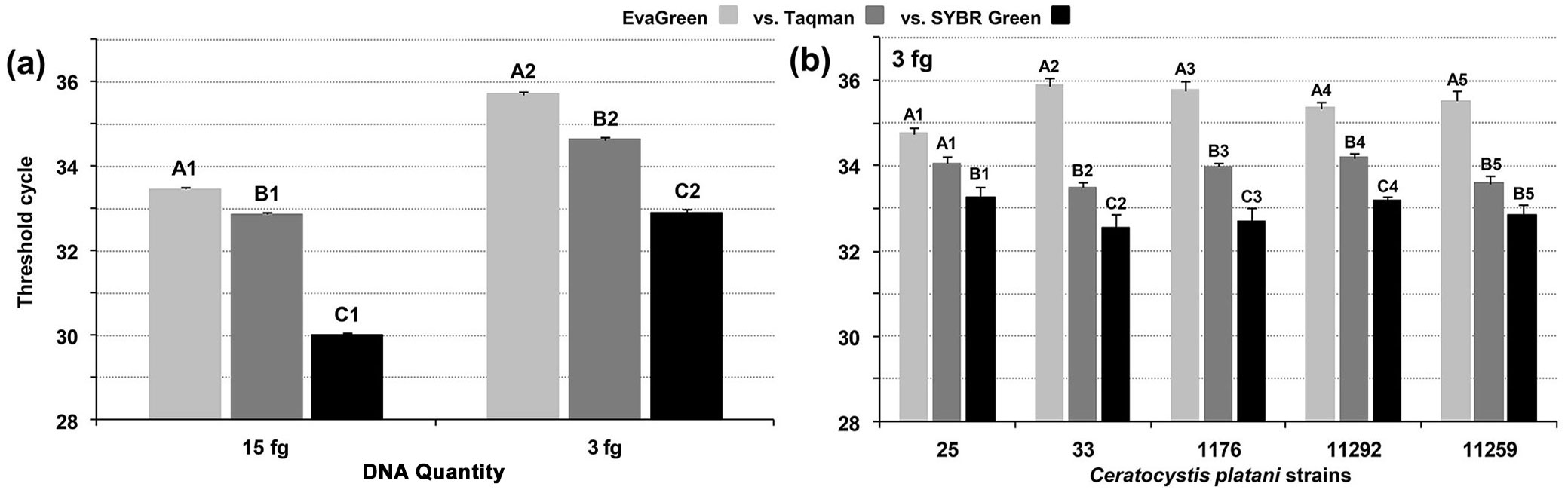
Testing five additional C. platani strains (8-value dataset) confirmed the repeatability of ASe, i.e., the ability to detect 3 fg C. platani gDNA per PCR reaction (Fig. 3b, Tab. S4a). For each strain, the Ct mean of the detection limit was always the lowest using SYBR Green, the highest with EvaGreen and intermediate with Taqman, with significant differences in most cases (Fig. 3b, Tab. S1).
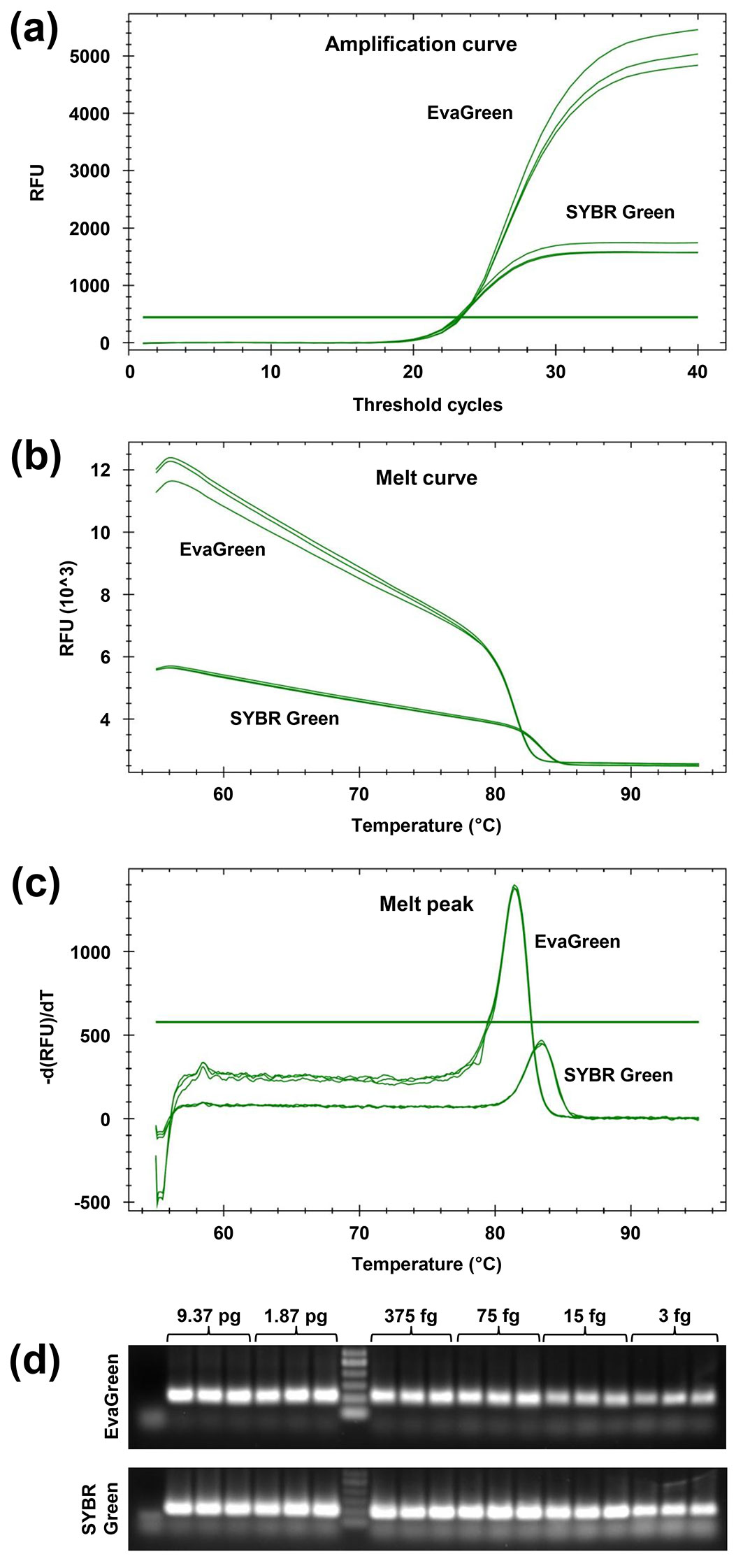
Comparing the two assays based on DNA-binding dyes, the fluorescent signals obtained during the cycling were different in intensity, as EvaGreen yielded a fluorescence that reached a plateau at or just above 5000 RFU (relative fluorescence units) and SYBR Green at 1800 or just below 2000 RFU (Fig. 4a). In line with this finding, a larger decrease in fluorescence in the melt curve, and a higher melt peak, were always recorded with EvaGreen compared to SYBR Green (Fig. 4b, Fig. 4c).
Fig. 4 - Comparisons between EvaGreen and SYBR Green assays. (a) Intensity of the fluorescent signals obtained on an equal quantity of template (RFU: relative fluorescence units). (b) The melt curves derived from the dissociation run. (c) The melt peaks. (d) Validation for using the cycling protocol in conventional PCR: agarose gel analysis of amplification products obtained from five-fold serially diluted gDNA of Ceratocystis platani, including 3 fg, i.e., the DQP-Ase. Amplicons are compared with a GeneRuler 50 bp DNA ladder (Thermo Fisher Scientific, Waltham, MA, USA).
We used the Taqman assay to assess different extraction events by different operators and different modalities of the DNA extraction procedure (scraping mycelium from agar or from cellophane). These experiments confirmed the repeatability of the Real-Time PCR and its capacity to detect 15 and 3 fg gDNA when varying the parameters described above (Fig. 5a, Fig. 5b, Fig. 5c). The mean Ct values at which detection occurred did not differ significantly between the extraction events or extraction modalities (Fig. 5a, Fig. 5b, Tab. S1), except that the mean Ct values of the agar modality were slightly lower than the cellophane modality at both 15 and 3 fg. This difference was statistically significant for 3 fg with a 99% confidence level (Fig. 5c), whereas the difference was significant for 15 fg with a 95% confidence level (data not shown).
Fig. 5 - Validation of the detection limits (in-mycelium DQP-ASe) in relation to different gDNA extraction events performed by different operators (i.e., reproducibility) and different modalities of collecting the fungal mycelium for DNA extraction: the agar modality (agar) and the cellophane modality (cell.). (a) Ct mean of each experiment performed by a single operator using gDNA extracted by operator A and C, each using the two collection modalities (8-value dataset for each mean). (b) Comparison of the various extraction events by different operators (A and C; 24-value dataset for each mean). (c) Agar modality vs. cellophane modality (48-value dataset for each mean). The Taqman assay was used for this validation (99.0% confidence level).
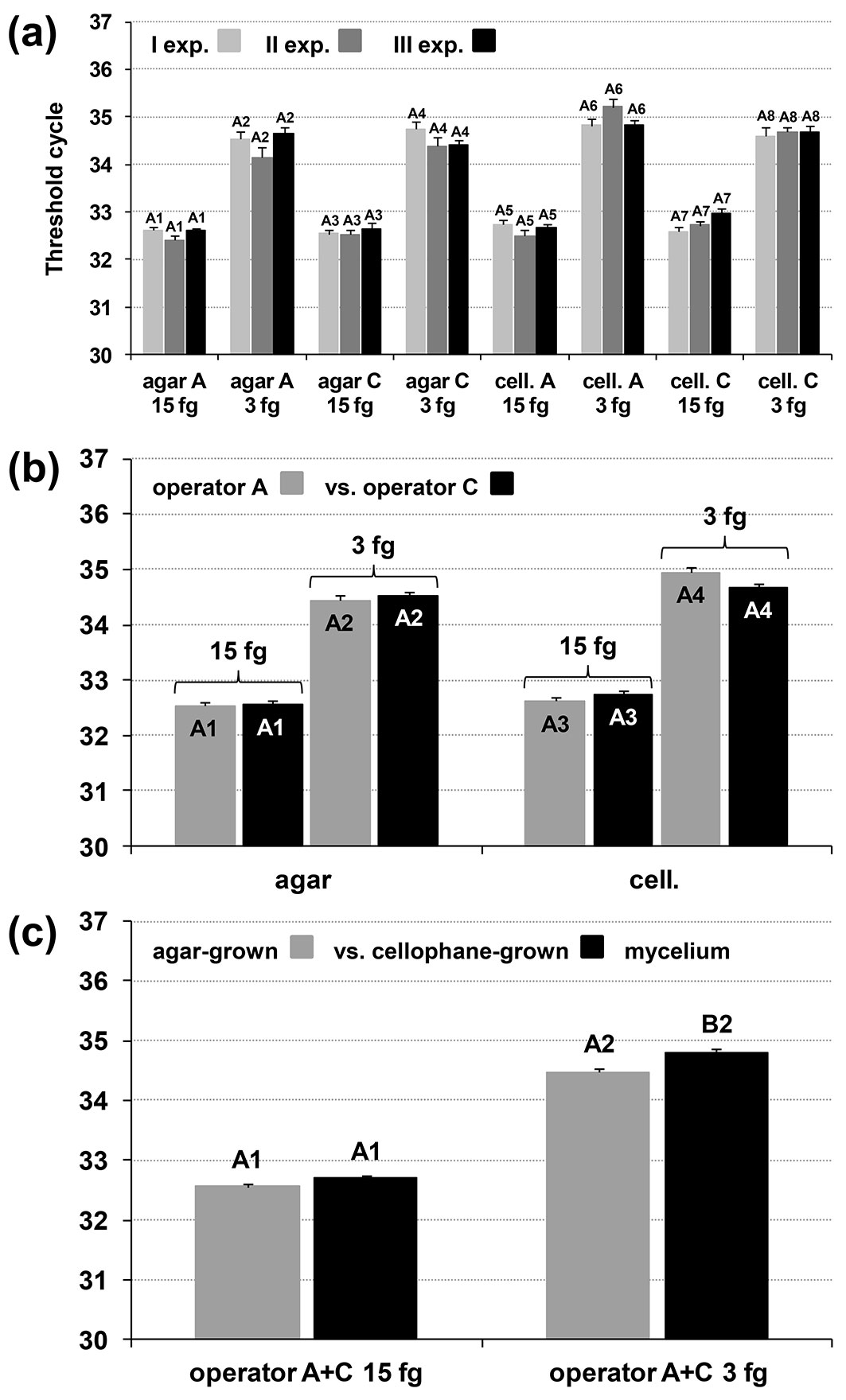
In all the experiments described above, negative and positive control reactions responded as expected, confirming the correctness of the Real-Time PCR run.
Melting temperature of target amplicon and agarose gel analyses
No significant difference was detected in the melting temperature of the amplicon obtained from extracted fungal gDNA and from the plasmid containing the PCR target (Tab. S1 in Supplementary material). The melting temperature was 81.35 °C (SD: 0.05) in the EvaGreen assay and 83.43 °C (SD: 0.06) in the SYBR Green assay. This difference in the melting temperature was highly significant at the 99% confidence level (Fig. 4c, Tab. S1).
Amplification products derived from EvaGreen and SYBR Green Real-Time PCR reactions that were charged with 9.37 pg to 3 fg gDNA (five-fold dilution series) template were all clearly detectable in agarose gel analysis (Fig. 4d). For a further confirmation of the utility of agarose gel analysis, EvaGreen and SYBR Green amplification products from four naturally-infected trees were also electrophoresed. Three of these were derived from samples that were detected at late Ct, but all the amplicons were clearly detectable in agarose gel analysis (Fig. S2 in Supplementary material). The agarose gel analysis also showed that EvaGreen and SYBR Green affected the electrophoretic mobility of the amplicons differently (Fig. S2).
Validation of in-necrotic-wood DQP and DQS-ASe, DSe and DSp
Spiking experiments showed that the Real-Time PCR using extractions from necrotic wood uninfected by C. platani were still capable of detecting the added 3 fg C. platani gDNA. At this detection limit, the method was fully repeatable both qualitatively and quantitatively (no significant differences in the mean Ct values - Fig. 6, Tab. S4b). These results suggested that the in-necrotic-wood DQS-ASe of the Real-Time PCR SYBR Green assay is 0.5 fg µl-1 when 6 µl of extracts of necrotic wood are added to the reaction.
Fig. 6 - Validation of the detection limits in necrotic wood with spiking experiments (in-necrotic-wood DQP and DQS ASe). PCR reactions were spiked with 3 fg Ceratocystis platani gDNA and 6 µl meDNA extracted from necrotic wood of plane trees not affected by canker stain. Letters indicate homogeneous groups (99.0% confidence level). Vertical lines represent the standard error of the mean.
Both the 6 µl and 2 µl volume of extracts from samples collected from trees artificially (15 samples) and naturally (28 samples) infected with C. platani were positively detected with all three assays. The 6 µl volume led to a positive detection at a lower Ct than the 2 µl volume in most cases. The exception was with artificially-infected samples that were dead since long time before sampling, which gave a false negative detection in most cases when using the 6 µl volume dose. Generally, the mean Ct at which C. platani was detected was the lowest in SYBR Green compared with EvaGreen and Taqman assays (Tab. S5 in Supplementary material).
A summary of Real-Time PCR detection limits for different types of extracted DNA and analytical sensitivity is reported in Tab. 2.
Tab. 2 - Real-Time PCR detection limits for different types of extracted DNA and analytical sensitivity (Ase). (DQP): DNA quantity per PCR reaction; (DQS): DNA quantity in the sample to be tested. In-mycelium means that the detection limits were validated using Ceratocystis platani gDNA extracted from a pure fungal culture; in-necrotic-wood refers to detection limits validated using meDNA extracted from necrotic wood of trees infected with Fomitiporia mediterranea and uninfected with C. platani; TS is the tested sample. Values of in-mycelium ASe refers to EvaGreen, Taqman and SYBR Green assays; values of in-necrotic-wood ASe refers to SYBR Green assay.
| ASe parameters | In-mycelium DQP-ASe |
In-mycelium DQS-ASe |
In-necrotic-wood DQP-ASe |
In-necrotic-wood DQS-ASe |
|---|---|---|---|---|
| Extracted DNA | Tested on gDNA extracted from a pure fungal culture |
Tested on gDNA extracted from a pure fungal culture | Tested on meta DNA extracted from necrotic wood of plane tree infected with F. mediterranea and spiked with C. platani gDNA | Tested on meta 6 μl DNA extracted from necrotic wood of plane tree infected with F. mediterranea plus 3 fg C. platani gDNA |
| Detection limits by Real-Time PCR | 3 fg per PCR reaction | 1.5 fg µl-1 TS if 2 µl of extract added; 0.375 fg µl-1 TS if 8 µl of extract added | 3 fg per PCR reaction whether 2 (data not shown) or 6 µl extract of necrotic wood added | 0.5 fg µl-1 TS if 6 µl of extract added |
In the DSp experiments, the 23 isolates representing the species and strains within the Ceratocystidaceae showed a wide range of sensitivities to both the EvaGreen and Taqman assays, but the two assays gave very similar Ct values for each of the tested concentrations (Tab. 1). Nine of the species yielded no signal at the highest gDNA dose tested. Six species gave only weak signals, i.e., detected at very late Ct with high gDNA levels (500 pg or 5 ng per reaction). Eight isolates representing species and strains of the genus Ceratocystis were detected at 5 fg to 50 pg per reaction. Most of these species are found in the Latin American Clade (LAC) of the genus, which includes C. platani, though C. ficicola of the Asian-Australasian Clade (AAC) and C. variospora of the North American Clade (NAC) were detected at 50 pg per reaction (Tab. 1). Among the LAC members, C. colombiana and the sweet potato (Ipomea batata) strain of C. fimbriata were detected at 50 pg; C. fimbriata strains from coffee (Coffea sp.) and Eucalyptus sp. were detected at 50 fg; and C. cacaofunesta and a mango (Mangifera indica) strain of C. fimbriata were detected at 5 fg per reaction.
In all the experiments described above, negative and positive control reactions responded as expected, confirming the correctness of the Real-Time PCR run.
Discussion
In-depth validation and implementation of the Real-Time PCR method developed by Pilotti et al. ([24]) follows the EPPO guidelines for validation of diagnostic methods ([18], [20]). We established the different ways in which analytical sensitivity (ASe) can be defined and determined the diagnostic potential of the method and its versatility (see the flow chart in Fig. S3, Supplementary material). The analyses validated the specificity of the primers and showed the high analytical and diagnostic sensitivity of the Real-Time PCR assays.
The five-fold dilution series and 8-replication-based experiments highlighted that the assays repeatedly detected gDNA extracted from mycelium at concentrations as low as 3 fg per PCR reaction, regardless of whether the agar or the cellophane collection procedures were used or whether different operators had performed the DNA extraction and Real-Time PCR. A detection limit of 0.5 fg µl-1 in extracts from necrotic wood was established by spiking 6 µl of wood extracts with 3 fg gDNA, and this enabled a refinement of the previously declared ASe values ([19] - Tab. 2). Any comparison with ASe of method by Luchi et al. ([12]) would be rigorous only within an inter-laboratory comparison.
Scraping mycelium directly grown on PDA yielded gDNA preparations that worked as well or better than collecting mycelium from cellophane, suggesting that agar media did not inhibit the PCR reactions, in contrast to the findings of Gibb & Wong ([4]). In fact, extracts from agar media had slightly lower Ct values than extracts from cellophane. Moreira ([15]) has been reported that blocks of agarose containing embedded DNA improved the PCR amplification from templates naturally contaminated with powerful PCR inhibitors such as polysaccharides or humic acids. Thus, although agar may be an inhibitor of PCR ([4]), it may favor the removal of negligible quantities of PCR inhibitors, such as those released by fungal mycelium during DNA extraction.
Use of SYBR Green is a new technical feature and increases the versatility of the assay. Its validation meets the request by the Italian phytosanitary services. Interestingly, although in-mycelium DQP-Ase with SYBR Green was the same as in the EvaGreen and Taqman assays (3 fg per PCR reaction), the SYBR Green assay yielded an earlier signal when the template quantity was the same. The capacity of SYBR Green to confer earlier detection signals than the other assays was especially useful in confirming tests of tree samples that yielded late fluorescent signals. Thus, SYBR Green may be the best choice in some applications, especially compared to EvaGreen, and it could be best applied to samples suspected to contain an extremely low titer of target DNA.
Key benefits of a DNA dye for Real-Time PCR are its capacity to produce a robust fluorescence signal, thus giving amplification curves that rise for several cycles before reaching a plateau, and a large and rapid fluorescence fall following amplicon melting (a high and narrow melt peak) which enables detection of even single-base pair mismatches ([28]). A robust fluorescence signal depends on the ability of the dye to be used at a relatively high concentration and its preferential fluorescence response to dsDNA over ssDNA.
Mao et al. ([13]) established that EvaGreen can be used at four times higher concentration than SYBR Green (i.e., 1.33 µM vs. 0.34 µM) without causing a significant Ct delay or nonspecific product formation. At these concentrations, EvaGreen gave 4-5 times more fluorescence than SYBR Green. In addition, Mao et al. ([13]) found that increasing the SYBR Green concentration, shortening the extension time, and increasing the amplicon size caused a delay in reaching the detection Ct level, the formation of non-specific amplification products, and some loss of repeatability. Conversely, amplification reactions using EvaGreen showed a significant Ct delay, but only under much more challenging conditions than those sufficient to have a negative impact on the SYBR Green assay. Taken together the results of Mao et al. ([13]) suggest that EvaGreen should generally perform better than SYBR Green in Real-Time PCR, and should be more suitable for fast cycling protocols.
Our study confirmed the increased strength of the fluorescence signal of EvaGreen compared with SYBR Green. On the other hand, although our cycling protocol is very rapid, the SYBR Green assay compared to the EvaGreen assay was not subject to Ct delay, non-specific product formation, or loss of repeatability. There was no apparent primer dimerization in the SYBR Green assay in no-template reactions, even when lowering the annealing/extension temperature. In addition, the SYBR Green assay proved to be slightly, but significantly, more sensitive than the EvaGreen and Taqman assays. We hypothesize that the high performance of the SYBR Green assay can be explained by the effective optimization of the reaction mixture, which is also known to affect the performance of the Real-Time PCR fluorophores ([13]). The SYBR Green Supermix (BIO-RAD) that we used is specifically recommended for robust performance with demanding templates under challenging conditions.
The melting temperature of the target amplicon was higher when analyzed with SYBR Green compared with EvaGreen, with a gap of two degrees, in accordance with findings by Mao et al. ([13]). The 0.1 °C stepped melt rate is known to increase the peak resolution of amplicons that melt at similar temperatures, compared with peak resolutions based on wider steps, for which the risk of ambiguity is higher ([26]). Adopting the 0.1 °C stepped melt rate enabled to confirm the specificity of the primers and rule out the promotion of non-specific amplification with SYBR Green. In conclusion, we show that SYBR Green tested with a commercial supermix performs as well as or better than EvaGreen, and thus it represents a valuable new technical option at a lower cost.
Interestingly, the agarose gel analysis enabled visualization of the amplicon bands, even those derived from the lowest detectable template DNA in the PCR reaction, i.e., 3 fg gDNA. This confirms the DQP-Ase and highlights that this Real-Time PCR method could also be used in qualitative PCR by laboratories that are not equipped with a quantitative thermo-cycler. Indeed, the validation of this technical option was specifically requested by some Italian phytosanitary services.
We considered ASe in relation to pure fungal gDNA vs. meDNA extracted from necrotic wood and to different template volumes (2 or 6 μl of the extract in the reaction mixture). Indeed the nature of tissue source and the template volume are crucial for methods aimed at in-wood detection. Spiking experiments and diagnostic sensitivity (DSe) tests showed that the necrotic nature of the wood does not significantly affect the detection of C. platani gDNA in wood compared to gDNA from fungal mycelium (Fig. 6, Tab. S5). The fact that the PCR reaction can be challenged with extraction volumes greater than the canonical 2 μl (i.e., 6 μl) without the risk of inhibition means that the method is able to detect extremely low titers of C. platani gDNA in sapwood samples, namely extractions with as low as 0.5 fg µl-1 (in-necrotic-wood DQS-ASe). The ASe for in-necrotic-wood colonized by F. mediterranea should be considered close to the ASe of the assays in canker stain-affected wood, but the inhibitive power of the necrotic wood colonized by F. mediterranea might differ somewhat from discolored wood affected by canker stain.
The robustness of the Real-Time PCR was further highlighted by the DSe validation, in which 28 samples from trees naturally infected by C. platani were tested. The detection results clearly showed that the three assays work efficiently on wood samples colonized by C. platani. Use of 6 µl of wood extraction rather than 2 µl as template generally resulted in positive detections at earlier Ct values, suggesting that increasing template volume increases the detectable PCR target without significantly increasing putative PCR inhibition. However, extractions from necrotic wood that had been dead for a long time appeared to have a more inhibitory effect than extractions from freshly-killed tissue. This inhibitory effect was seen at the 6 µl volume dose but not at the 2 µl volume dose. The overall results suggest that the quantity of inhibitors contained in C. platani-infected wood samples can vary greatly according to a number of known and unknown parameters. The 2 µl volume dose should be considered routine, whereas 6 µl can be used when samples affected by the characteristic canker stain symptoms are negatively detected or detected with late fluorescent signals.
In several samples from naturally-infected trees, the Ct values were higher than 31, whereas in the previous detection experiments on naturally-infected trees only 4 out of 20 samples were detected at such high Ct values ([24]). The difference can be explained with our intentional lack of supervision during the sample collection phase in the current study. In general, the samples yielding high Ct values appeared to be collected from long-dead trees. Interestingly, only a few samples were positively detected with the microscope assay. This confirms previous results with artificially-infected trees ([24]), in which the effectiveness of detection by microscope was lower than by Real-Time PCR in wood that had been dead for a long time.
We investigated the specificity of the method among closely related and more distantly related species in the Ceratocystidaceae, and we obtained non-specific signals with some of the species and strains most closely related to C. platani. It was anticipated that the Taqman probe assay would be more specific than the EvaGreen assay, but that was not evident in the data. The strength of the reaction was strongly correlated with the concentration of fungal gDNA.
With most of the species tested, there was no or only limited detection at the highest concentration tested (5 ng per reaction). The genus Ceratocystis is now restricted to what was previously known as the C. fimbriata complex ([2]), and the eight isolates that were detected at 500 pg per reaction or less would now be considered Ceratocystis sensu stricto. Of these isolates, six reside in the Latin American Clade (LAC) of the complex with C. platani ([6]). There are numerous short regions of insertions and deletions in the ITS sequences of Ceratocystis, and these indels are not phylogenetically informative ([7]). However, they likely influence the specificity of the assays. Thus, it is not too surprising that genetic relatedness of the LAC strains to C. platani did not completely correspond to level of detection in the C. platani assays. Of the species tested, C. platani is most closely related to C. colombiana ([11]), which was detected only at 500 pg per reaction, while the tested isolate of C. cacaofunesta and a Brazilian isolate of C. fimbriata from mango were detected at just 5 fg per reaction. The target sequences of the latter two species differ at only the 5’ end of the forward primer of the assays, while the ITS sequence of C. colombiana differs at the target sites of both the primers and the probe.
The primers and probes for the C. platani assay could be modified to be adapted for specificity to other species in the family. The ITS1 region utilized in this assay is more variable among members of the LAC than the ITS2 region ([7]), and ITS1 is to be considered a good target. Luchi et al. ([12]) reported that their ITS2 assay did not cross react with other members of Ceratocystis, including a number of isolates that tested positive in our ITS1 assay. However, as they did not report the template concentration in the reactions for DSp experiments, their results are not comparable with ours. Moreover, the ITS2 target sights ([12]) of seven NCBI GenBank accessions of Ceratocystis spp. in the LAC are identical to those of C. platani, and we would expect lack of specificity with the ITS2 assay by Luchi et al. ([12]) (data not shown). Indeed complete specificity within Ceratocystidaceae seems difficult to realize for any method targeted on ITS region.
Nonetheless, the ITS1 assay employed here is specific for the purpose of detecting C. platani. Of the other members of the Ceratocystidaceae within the geographic distribution of C. platani in Europe and Asia Minor ([25]), Endoconiophora coerulescens and E. resinifera were detected occasionally at 5 ng or 500 pg per reaction, but these species are only known in wood of Pinaceae ([5]). There may be some geographic overlap for C. harringtonii or C. variospora with C. platani in the USA ([8]), but these and the other tested species are not known to colonize Platanus spp., and C. platani is highly specific to this host ([1], [3]).
Problems of specificity with Ceratocystis sensu stricto members could be overcome by integrating other technologies. A minor groove binder (dihydrocyclopyrroloindole tripeptide) conjugated at the 3’ end of the Taqman probe hyper-stabilizes the pairing of the probe on the target template, enabling the increase in Tm of short probes, which in turn increases specificity ([10], [12]). As our probe and primers are long (28 and 26-mer), a more suitable choice could be the inclusion of locked nucleic acids (LNA) bases at nucleotide positions that distinguish C. platani from the other strains/species. LNA bases are high-affinity RNA analogs in which an oxymethylene bridge locks the ribose ring in the ideal conformation for specific binding to the complementary base. This increases hybridization stability and ensures discrimination between closely related sequences down to as little as one nucleotide difference ([9]).
Conclusions
A high-performing method for detection of C. platani is essential to safeguard those countries that remain unaffected by canker stain. The in-depth validation of the method according to EPPO standards guarantees users of its effectiveness and promotes its utilization. The analytical sensitivity and the diagnostic sensitivity of the method were thoroughly investigated, implemented and validated according to stringent intra-laboratory repeatability and reproducibility conditions. The method proved to be highly robust, effective and multi-faceted. Moreover, the exploitation of the different types of analytical sensitivities is worthy of special attention as it helps in defining guidelines on how the various laboratories that validate diagnostic methods could collaborate through inter-laboratory and proficiency tests.
This work paves the way for a third conclusive step, a test performance study through an inter-laboratory comparison.
List of abbreviations
The following abbrevations have been used throughout the text:
- DQP-ASe: DNA quantity per PCR reaction - Analytical Sensitivity;
- DQS-ASe: DNA quantity in the sample to be tested - Analytical Sensitivity;
- TS: tested sample;
- DSe: diagnostic sensitivity;
- DSp: diagnostic specificity.
Acknowledgements
VL, VM and AB performed all DNA extractions and Real-Time PCR; GDL and MP, performed specimen preparation and microscopic observation for Ceratocystis platani detection; CLW performed DNA extractions of Ceratocystidaceae and Real-Time PCR for DSp; TCH made available the Ceratocystidaceae collection, conceived DSp experiments and revised the manuscript; MP conceived the study, performed statistical analyses and drafted the manuscript. All the authors were responsible for drafting the manuscript. This research was partly supported by the MIPAAF (the Italian Ministry of Agricultural and Forestry Policies): “Azioni a supporto della protezione delle piante (ASPROPI)”.
References
Online | Gscholar
Gscholar
Authors’ Info
Authors’ Affiliation
Vanessa Modesti
Angela Brunetti
Giovanni Di Lernia
Massimo Pilotti
Research Centre for Plant Protection and Certification (CREA-DC), Consiglio per la ricerca in agricoltura e l’analisi dell’economia agraria, v. C. G. Bertero 22, I-00156 Rome (Italy)
Thomas C Harrington
Iowa State University, Department of Plant Pathology and Microbiology, 1344 ATRB, Ames, IA 50011 (USA)
Corresponding author
Paper Info
Citation
Lumia V, Modesti V, Brunetti A, Wilkinson CL, Di Lernia G, Harrington TC, Pilotti M (2018). Real-Time PCR for Ceratocystis platani detection: in-depth validation to assess the diagnostic potential and include additional technical options. iForest 11: 499-509. - doi: 10.3832/ifor2527-011
Academic Editor
Alberto Santini
Paper history
Received: Jun 19, 2017
Accepted: May 15, 2018
First online: Jul 18, 2018
Publication Date: Aug 31, 2018
Publication Time: 2.13 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2018
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 51374
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 42605
Abstract Page Views: 3753
PDF Downloads: 3934
Citation/Reference Downloads: 6
XML Downloads: 1076
Web Metrics
Days since publication: 2935
Overall contacts: 51374
Avg. contacts per week: 122.53
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Jul 2026)
Total number of cites (since 2018): 12
Average cites per year: 1.33
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Feasibility study of near infrared spectroscopy to detect yellow stain on cork granulate
vol. 11, pp. 111-117 (online: 31 January 2018)
Research Articles
Outcome of Ceratocystis platani inoculations in Platanus × acerifolia in relation to season and inoculum dose
vol. 9, pp. 608-617 (online: 17 March 2016)
Short Communications
Detection and quantification of the air inoculum of Caliciopsis pinea in a plantation of Pinus radiata in Italy
vol. 12, pp. 193-198 (online: 10 April 2019)
Research Articles
Latent infection of Biscogniauxia nummularia in Fagus sylvatica: a possible bioindicator of beech health conditions
vol. 9, pp. 49-54 (online: 18 June 2015)
Research Articles
Lenticel infection in Fraxinus excelsior shoots in the context of ash dieback
vol. 12, pp. 160-165 (online: 04 March 2019)
Research Articles
Hearing nature’s heartbeat: towards large-scale real-time forest monitoring network in Italy
vol. 18, pp. 202-211 (online: 09 August 2025)
Research Articles
Identification of the ambrosia beetle Anisandrus dispar (Fabricius) (Coleoptera Curculionidae Scolytinae) using TaqMan™ probe assay on biological samples
vol. 16, pp. 182-187 (online: 30 June 2023)
Research Articles
Use of unmanned aerial vehicles for the diagnosis of parasitic plant infestation at the crown level in Pinus hartwegii
vol. 16, pp. 282-289 (online: 28 October 2023)
Research Articles
The concept of green infrastructure and urban landscape planning: a challenge for urban forestry planning in Belgrade, Serbia
vol. 11, pp. 491-498 (online: 18 July 2018)
Research Articles
Forest therapy in Italy: proposal of a standard procedure for validation of suitable sites
vol. 17, pp. 192-202 (online: 04 July 2024)
iForest Database Search
Search By Author
Search By Keyword
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords
PubMed Search
Search By Author
Search By Keyword







