Patterns of genetic diversity in European beech (Fagus sylvatica L.) at the eastern margins of its distribution range
iForest - Biogeosciences and Forestry, Volume 10, Issue 6, Pages 916-922 (2017)
doi: https://doi.org/10.3832/ifor2446-010
Published: Dec 10, 2017 - Copyright © 2017 SISEF
Research Articles
Collection/Special Issue: COST Action FP1202
Strengthening conservation: a key issue for adaptation of marginal/peripheral populations of forest trees to climate change in Europe (MaP-FGR)
Guest Editors: Fulvio Ducci, Kevin Donnelly
Abstract
Populations located at the periphery of the species’ distribution range may play an important role in the context of climate change. These peripheral populations may contain specific adaptations as a result of extreme environmental conditions. The aim of this paper was to assess within population genetic diversity and among population differentiation in one of the most important forest tree species in Europe, European beech (Fagus sylvatica), at the eastern margins of its natural range. We analysed four peripheral, isolated populations and five core populations from the continuous natural range along the Carpathian Mountains using a set of microsatellite markers. Higher levels of genetic diversity as measured by allelic richness (7.34 vs. 6.50) and observed heterozygosity (0.71 vs. 0.59) were detected in core populations than in peripheral ones. Population differentiation was slightly higher among peripheral populations than among core, Carpathian populations. There was strong evidence of bottleneck effects in two out of the four peripheral, isolated populations. Both core, Carpathian populations and peripheral, lowlands populations share the same chloroplast haplotype suggesting a common geographical origin from the putative Moravian refuge area. Past long distance founding events with material from the Carpathian mountain chain might explain the occurrence of small, isolated beech populations towards the steppe in the south-east of Romania. Our genetic data may contribute to a better understanding of the evolutionary history of the remnants of beech scattered occurrences at the eastern margins of species’ distribution range.
Keywords
Fagus sylvatica, Genetic Diversity, Peripheral Populations, Bottleneck Effect
Introduction
Populations residing at the current low-latitude and low-altitude margins of species’ distribution range are particularly important in the context of climate change ([3], [26]). Peripheral populations, which currently face higher risks of extinction, may play a critical role in determining species responses under climate change ([19], [26]). These peripheral populations are typically smaller and isolated from the continuous distribution range of the species and, as a result, are likely to experience increased genetic drift and to receive less immigrants than core populations ([4]). Moreover, peripheral populations at the warmer margins of the species distribution may experience higher selection pressure exerted by the warmer climate and thus harbor valuable adaptations. Lower neutral genetic variation as well as higher differentiation rates are expected in peripheral, isolated populations than in core populations from the continuous distribution range ([15]). In addition to climate change, peripheral tree populations may be affected by human activities, such as browsing by cattle, deforestation, and improper forest management.
So far, there are a limited number of studies on genetic diversity of peripheral versus core populations of forest tree species. Significantly higher allelic and genotypic diversity in peripheral populations than in core populations was reported in Eastern white pine ([5]). However, similar values for heterozygosity across peripheral and core populations were observed in the same study as well as in Scots pine ([43]), eastern white cedar ([33]) and Sitka spruce ([22]). Evidence of bottlenecks was reported in peripheral populations of Sitka spruce but not in Scots pine populations. Generally, the differences between peripheral and core populations as estimated using neutral genetic markers were statistically significant only for certain diversity indices which vary from one study to another.
In this study, we focused on common beech (Fagus sylvatica), one of the most important forest trees in Europe. Common beech occupies about 21 million hectares ([21]) and plays a crucial role for the society, economy and environmental health. It is also a keystone species that fulfills central functions within its ecosystems and interacts with hundreds of associated plant, animal and fungal species. At present, it is the most common forest tree species in Romania, occupying approximately 33% of the forest area and representing 40% of the growing stock ([2]). Unlike in western and central Europe, beech was not planted for forestry purposes in Romania and, consequently, the current populations are autochthonous ([40]). Only a few studies included samples of beech from Romania ([8], [25], [29]) and no investigation was done on peripheral, small, disjunct populations from the south-eastern part of the country where European beech reaches the eastern edge of its range ([40]). The most isolated peripheral beech population is located close to the Danube Delta, in the old Macin Mountains (maximum altitude: 467 m a.s.l.) formed in the second part of Paleozoic, during the Hercynian orogeny. This population grows only along a deep valley and on a north facing slope, and might be of Tertiary origin ([23]). A Balkan origin of this particular beech population was also assumed ([10]). This is in contrast with the geographical origin of existing beech populations in the nearby Carpathian Mountains, which may originate from the Moravian refugium. A secondary refugium might have been in Apuseni Mountains (western Romania), but did not contribute to the colonization of the Carpathians ([29]).
Understanding the evolutionary history of these peripheral beech populations, which are currently threatened by climate change and human activities, can help undertake appropriate management and conservation measures.
In this study, we assessed the genetic diversity of European beech at its eastern margins of the distribution range using a set of microsatellite markers. The specific questions of this paper are: (i) Is within population genetic diversity lower and population differentiation higher in peripheral, isolated populations than in core populations from the continuous Carpathian range? (ii) Is there any evidence based on chloroplast DNA variation of a different geographical origin of existing peripheral and core populations? (iii) Do peripheral, isolated populations show a significant bottleneck signature?
Material and methods
Study site and sampling
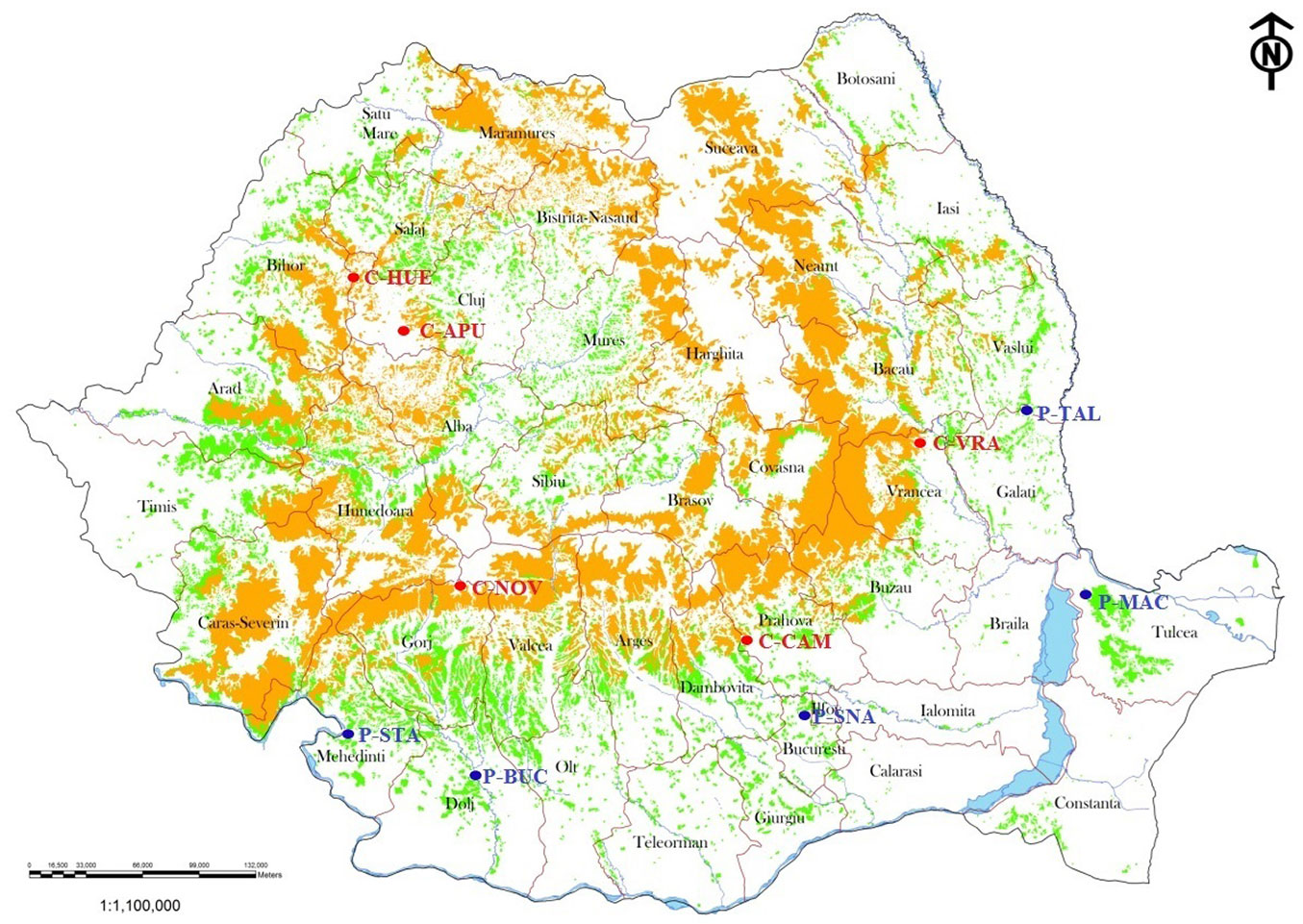
Our analysis included nine populations of European beech sampled throughout the natural range of the species in Romania (Fig. 1). The sampled populations were grouped into two categories (Tab. 1): four peripheral, isolated populations (P-MAC, P-SNA, P-STA and P-TAL) and five core populations (C-HUE, C-APU, C-NOV, C-CAM and C-VRA). The core populations are located on both sides of the South-Eastern Carpathian Mountains in the continuous natural range of beech. We also sampled the last four remaining living individuals from a small, isolated beech population (Bucovat, P-BUC) located in the lowlands of southern Romania (Fig. 1). Two peripheral populations (P-SNA and P-TAL) were sampled exhaustively. Material was collected from 50 individual trees in the remaining populations. The sampled individuals were at least 30 m apart in the populations that were not exhaustively sampled. A total of 462 individual trees were sampled in 2012. Only three randomly chosen beech individuals per population, except for population P-MAC with five individuals, were analyzed at chloroplast DNA level. The material consisting of buds or leaves was stored at -60 °C before further analyses.
Fig. 1 - Geographic location of the sampled beech populations. (P): peripheral; (C): core. Abbreviations and geographic coordinates of the populations are given in Tab. 1. (Orange area): continuous range of European beech in the Carpathian Mountains; (green area): oak forests.
Tab. 1 - Geographic location, sample size and climate conditions of the sampled beech populations. Sampling of all remaining trees was done in two populations (P-SNA and P-TAL). (P): peripheral; (C): core.
| Label | Population | No. of samples |
Latitude N | Longitude E | Altitude (m) |
Annual average temperature (°C) |
Annual rainfall (mm) |
|---|---|---|---|---|---|---|---|
| P-MAC | Macin | 100 | 45°16′ | 28°10′ | 125 | 10.5 | 479 |
| P-SNA | Snagov | 37 | 44°43′ | 26°09′ | 92 | 10.6 | 585 |
| P-STA | Stârmina | 50 | 44°29′ | 22°45′ | 77 | 10.4 | 591 |
| P-TAL | Talasmani | 21 | 46°07′ | 27°50′ | 230 | 9.4 | 538 |
| C-HUE | Huedin | 50 | 46°58′ | 22°43′ | 446 | 8.7 | 672 |
| C-APU | Apuseni | 50 | 46°40′ | 23°01′ | 1130 | 5.9 | 821 |
| C-NOV | Novaci | 50 | 45°13′ | 23°40′ | 560 | 8.3 | 702 |
| C-CAM | Câmpina | 50 | 45°06′ | 25°43′ | 520 | 8.3 | 673 |
| C-VRA | Vrancea | 50 | 46°05′ | 27°01′ | 315 | 9.6 | 559 |
Genotyping
DNA was extracted from buds or leaves using the cetyltrimethyl ammonium bromide (CTAB) method ([11]). DNA concentration and purity were determined spectrophotometrically with a Nanodrop 8000. Seven gSSRs originally developed for F. sylvatica (FS3-04, FS4-46, Mfs11 - [35], [42]) and F. crenata (Sfc0018, Sfc0161, Sfc1063 and Sfc1143 - [1]) and one EST-SSRs (Fir065) originally developed for Quercus spp. ([13]) were used. FS4-46 was excluded from further analysis because of some ambiguities in its interpretation and due to the presence of a large number of null alleles. Polymerase chain reaction (PCR) was carried out in Corbett and Eppendorf Thermal Cyclers. The amplification was performed in 10 μL of reaction mixture consisting of: 5× PCR Buffer (Promega), 0.2 mM each of dNTP, 2.5 mM MgCl2, 0.4-0.5 μM each of primers and 1.0 U of Taq DNA polymerase (Promega). The PCR profile was as follows: 15 minutes of initial denaturation at 95 °C followed by 30 cycles of 1 min denaturation at 94 °C, a 30 s annealing step at 47 °C (for multiplex 2 - Mfs11, Fir065, FS4-46 and Sfc1143) or 55 °C (for multiplex 1 - Sfc0018, Sfc0161, Sfc1063 and FS3-04), a 1 min elongation step at 72 °C and a 20 min final extension step at 72 °C.
Three polymorphic chloroplast microsatellites (ccmp-4, ccmp-7 and ccmp-10) were also amplified. The PCR reactions and performed in a 15 µL volume containing 5× PCR Buffer (Promega), 2 mM MgCl2, 0.2 mM dNTPs, 0.3 µM of each primer, 0.25 units of Promega Taq DNA polymerase. The PCR protocol consisted of one cycle of initial denaturation at 94 °C for 15 min, followed by 35 cycles (ccmp-7 and ccmp-10) or 30 cycles (ccmp-4) of denaturation at 94 °C for 1 min, annealing at 50 °C for 1 min, and extension at 72 °C for 1 min. A final extension cycle at 72 °C for 10 min followed. Amplified PCR products were diluted and were than run on a GemoneLab GeXP Genetic Analyser® using Frag-3 method and Size Standard 400. The products were further analyzed using Fragment Analysis Software using default parameters and PA ver. 1.0 dye correction.
Microsatellite markers were tested for genotyping errors due to large allele drop-out, scoring of stutter peaks and non-amplified alleles using Micro-Checker® ver. 2.2.0.3 ([41]). The software indicated the presence of null alleles at very low frequencies (less than 2%) for three markers (Sfc0018, Sfc0161 and Fir065) in only two populations (C-APU and C-NOV). No evidence of large allele drop-out or scoring of stutter peaks was found in the populations.
Genetic diversity and differentiation
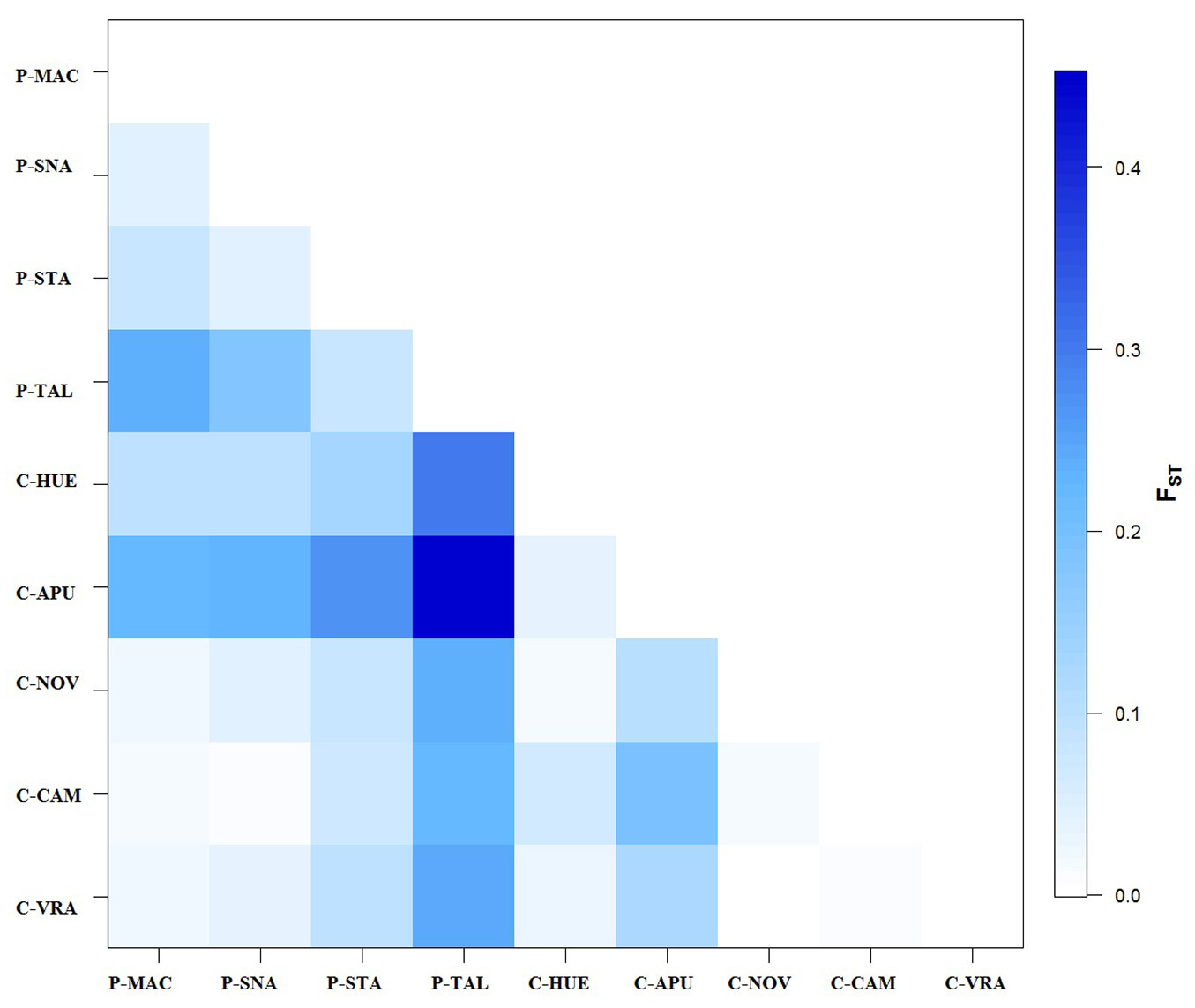
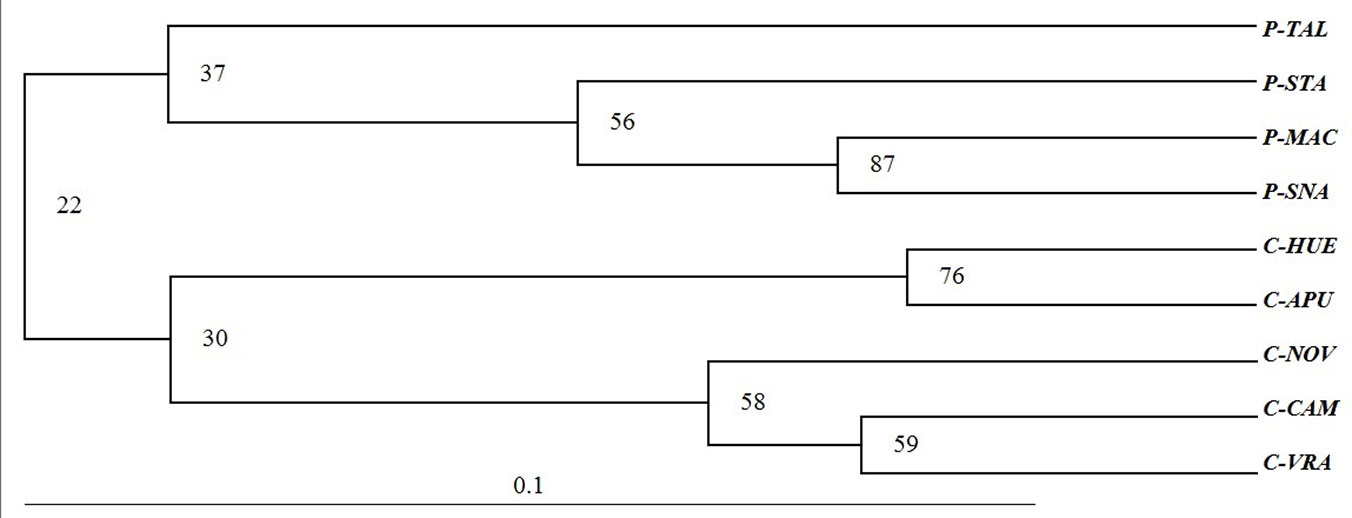
The software GenAlEx® ver. 6.5 ([36]) was used to estimate allele frequencies and standard genetic diversity indices: average number of alleles per locus (Na), effective number of allele (Ne), observed heterozigosity (Ho), expected heterozigosity (He) and fixation index (F). Allelic richness (AR), a measure that is independent of sample size, was estimated with FSTAT ver. 2.9.3 ([24]). Student’s t-test was used to examine differences between mean values of genetic diversity measures. Analysis of Molecular Variance (AMOVA) was performed using the software Arlequin® ver. 3.5.2.2 ([18]). A matrix of pairwise genetic differentiation measures between all populations pairs was computed. For genetic differentiation among beech populations, pairwise FST’s were computed using the same software. The significance of the FST statistics was tested by 10.000 permutations. The graphical representations of all pairwise FST were done using the Rfunction of the package “pairFstMatrix.r” ([39]) implemented in Arlequin via Rcmd (Fig. 2). An Unweighted Pair Group Method with Arithmetic Mean (UPGMA) clustering was computed with 100 bootstrap replications, based on Nei’s standard genetic distance ([30]) using the software Populations ver. 1.2.31 ([28]) and TreeView ver. 1.6.6 ([32]).
Fig. 2 - Matrix of pairwise genetic differentiation (FST) among beech populations.
Genetic assignment
The Bayesian clustering method implemented in the software STRUCTURE ver. 2.3.3 ([38]) was used to genetically assign individuals to clusters. Simulations were run for 100.000 steps following a burn-in period of 50.000 steps, considering values of K (number of clusters) from one to 10, with 3 replications for each value of K. The analysis was performed using admixture, correlated allele frequencies and no prior information on sampling location. The number of population clusters was estimated using ΔK parameter according to Evanno et al. ([17]) using the STRUCTURE HARVESTER program ([14]).
Bottleneck analysis
The software BOTTLENECK ([9]) was used to test for recent change in population size. We tested all beech populations for a bottleneck signature under the stepwise mutation model (SMM), infinite alleles model (IAM) and two phase model (TPM). We tested the significance levels using 1000 simulation iterations and both Wilcoxon’s signed rank test and standardized differences test.
Results
Within population genetic diversity
Eight to 22 alleles were observed per locus, with a total of 98 alleles across all populations and loci. Private alleles (6) were found in a single peripheral population (P-STA). Five private alleles were detected in the core populations, as follows: three in C-APU, one allele in C-HUE and one in C-VRA. Most of the private alleles except for one (Fir65-183bp) in population P-STA were rare (p<0.05). The values for the basic statistics of genetic diversity are listed in Tab. 2. Only two parameters showed significant differences (p< 0.05) between peripheral populations and core populations. Thus, the value of allelic richness (AR), a genetic parameter without population size bias, and observed heterozygosity (Ho) was higher for core populations than for peripheral ones. Gene diversity and fixation index showed similar values (Tab. 2).
Tab. 2 - Basic genetic statistics averaged across seven microsatellite loci for each population and group (peripheral and core). (N): sample size; (Na): mean number of alleles per locus; (Ne): number of effective alleles; (AR): allelic richness; (Ho): observed heterozygosity; (He): gene diversity; (F): heterozygote deficit; (SE): standard error.
| Population | N | Na | Ne | AR | Ho | He | F | |
|---|---|---|---|---|---|---|---|---|
| P-MAC | 85.7 | 9.14 | 4.61 | 6.697 | 0.686 | 0.718 | 0.041 | |
| P-SNA | 34.1 | 7.57 | 4.56 | 6.603 | 0.641 | 0.728 | 0.116 | |
| P-STA | 46.4 | 9.57 | 4.34 | 7.438 | 0.584 | 0.693 | 0.139 | |
| P-TAL | 19.1 | 5.57 | 2.58 | 5.273 | 0.433 | 0.491 | 0.116 | |
| C-HUE | 48.9 | 10.29 | 3.87 | 7.328 | 0.583 | 0.682 | 0.127 | |
| C-APU | 47.4 | 9.86 | 3.87 | 7.164 | 0.644 | 0.686 | 0.073 | |
| C-NOV | 49.0 | 9.57 | 5.23 | 7.788 | 0.734 | 0.752 | 0.029 | |
| C-CAM | 47.7 | 9.71 | 4.79 | 7.481 | 0.729 | 0.751 | 0.023 | |
| C-VRA | 49.1 | 9.14 | 4.87 | 7.076 | 0.771 | 0.728 | -0.047 | |
| Peripheral populations | Mean | 46.3 | 7.96 | 4.02 | 6.503 | 0.586 | 0.658 | 0.103 |
| SE | 0.6 | 1.15 | 0.81 | 0.107 | 0.071 | 0.073 | 0.055 | |
| Core populations | Mean | 48.4 | 9.71 | 4.53 | 7.367 | 0.712 | 0.720 | 0.041 |
| SE | 0.4 | 1.35 | 0.78 | 0.350 | 0.064 | 0.058 | 0.058 | |
| Overall | Mean | 47.5 | 8.94 | 4.30 | 6.983 | 0.645 | 0.692 | 0.069 |
| SE | 2.1 | 0.44 | 0.27 | 0.278 | 0.025 | 0.023 | 0.018 | |
Genetic differentiation among populations
The analysis of molecular variance (AMOVA) showed that the majority of the variance is within populations (Tab. 3). The differentiation among beech populations was moderate (FST=0.0978). AMOVA also indicated a slightly higher population differentiation among peripheral populations than among core populations (Tab. 3). The matrix of pairwise FST values (Fig. 2) revealed that differentiation between two populations of the same category (e.g., peripheral) was usually lower than differentiation between populations of different categories. The vast majority of the population pairs (more than 89%) were significantly differentiated from each other (p<0.001). The strongest differentiation was found between a core (C-APU) and a peripheral (P-TAL) population (FST=0.4526). No genetic differentiation was observed between pairs of neighboring peripheral (P-SNA and P-MAC) and core populations (C-VRA and C-MAC).
Tab. 3 - Analysis of molecular variance (AMOVA) at seven nuclear microsatellite loci. (a): All nine populations; (b): peripheral populations; (c): core populations; (df): degrees of freedom.
| Test | Source of variation | df | Sum of squares |
Variance components |
Percentage of variation |
Prob. |
|---|---|---|---|---|---|---|
| (a) | Among populations | 8 | 31.432 | 0.03628 | 9.77 | <0.001 |
| Within populations | 899 | 301.231 | 0.33507 | 90.23 | <0.001 | |
| (b) | Among populations | 3 | 42.349 | 0.13785 | 5.36 | <0.001 |
| Within populations | 406 | 893.919 | 2.43592 | 94.64 | <0.001 | |
| (c) | Among populations | 4 | 51.026 | 0.10389 | 4.13 | <0.001 |
| Within populations | 493 | 1187.683 | 2.40909 | 95.87 | <0.001 |
Population genetic structure
The dendrogram constructed using Nei’s genetic distances between pairs of populations revealed two main groups, one for peripheral and one for core populations, respectively. However, there was a small reliability of nodes (22) based on bootstrap resampling. The most south-eastern peripheral populations (P-MAC and P-SNA) and north-western core populations appear to be very similar (Fig. 3).
Fig. 3 - UPGMA based on Nei ([30]) genetic distance between beech populations.
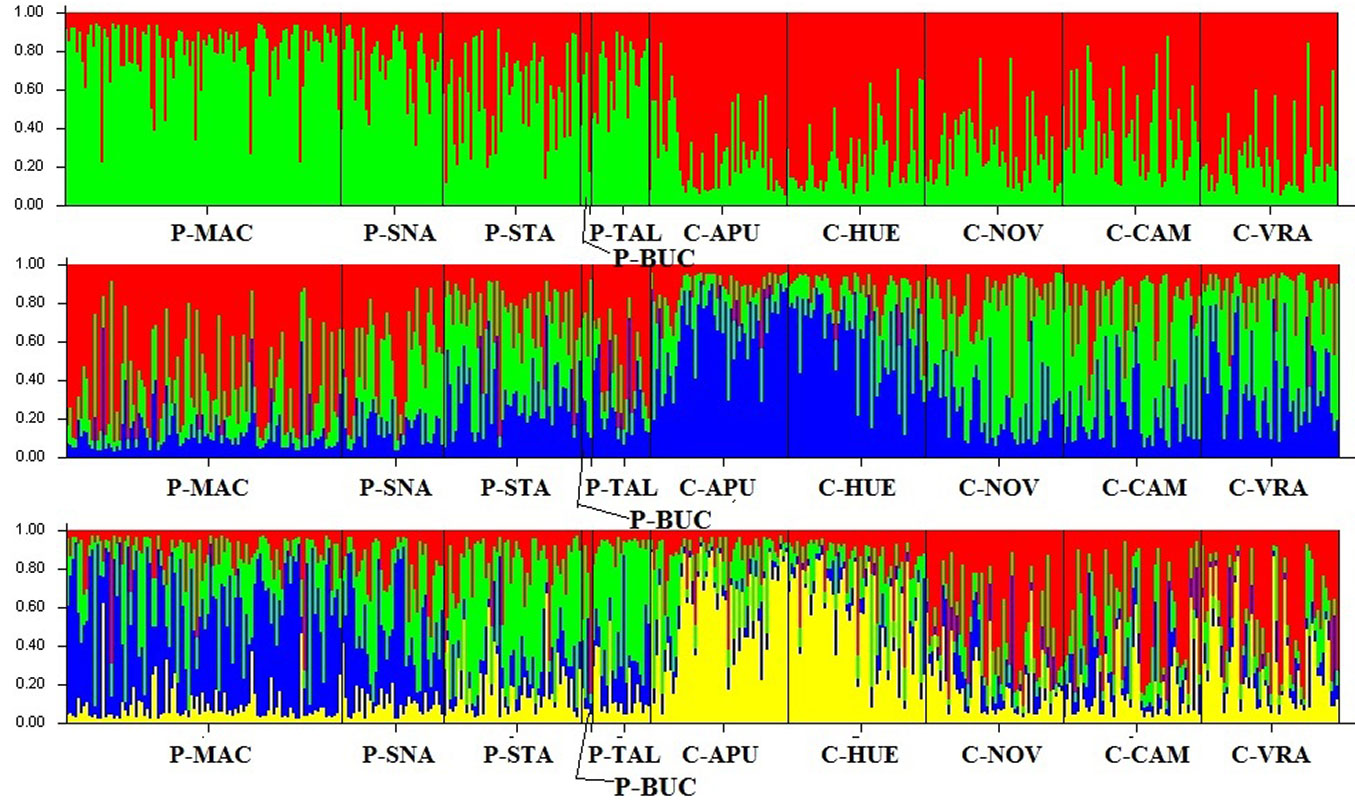
The most probable number of genetic clusters identified by the Bayesian analysis, using the ad hoc statistic ΔK, was four (additional data is given in Fig. S1 and Fig. S2 - Supplementary material). However, for K = 2, one cluster corresponds to peripheral populations (green color in Fig. 4) and one to core populations (red color). When having a third cluster (K = 3), peripheral populations remain together in one cluster while core populations are divided in two clusters in accordance with their geographic location: within Carpathian region, on the inner side of the South-Eastern Carpathian Mountains (blue color) and outside Carpathian ridge (green color). For K = 4, the initial group of peripheral populations is splitted into two clusters: the first cluster corresponds to populations P-MAC and P-SNA, which is in good agreement with the UPGMA dendrogram, and the second one to the rest of peripheral populations (Fig. 4).
Fig. 4 - Genetic structure revealed by seven microsatellite markers. Each individual tree is represented by a thin vertical line which is divided into color segments that are proportional to its membership in the genetic clusters (k=2, top panel; k=3, middle panel; k=4, lower panel) inferred in the Bayesian analysis. Membership values are averaged across three runs. Populations are separated by a thin black line. (P): peripheral; (C): core.
Chloroplast DNA analysis
The three chloroplast microsatellite makers were monomorphic in all populations. Allele 118bp, 146bp, and 109bp were observed at ccmp-4, ccmp-7, and ccmp-10, respectively. The same haplotype was observed at the chloroplast level across peripheral and core beech populations, revealing the same geographical origin.
Test for bottleneck signature
We observed evidence for recent bottlenecks or reductions in effective population size in two out of nine beech populations (Tab. 4). The two populations were both classified as peripheral: P-MAC under all three models (IAM, SMM and TPM) and P-SNA under the SMM model.
Tab. 4 - Test for recent bottlenecks in beech populations under stepwise mutation model (SMM), infinite alleles model (IAM) and two phase model (TPM). Significance was tested according to the Wilcoxon sign-rank test. Significant p-values are indicated with an asterisk (*).
| Population | SMM | IAM | TPM |
|---|---|---|---|
| P-MAC | 0.0016* | 0.0078* | 0.0016* |
| P-SNA | 0.0251* | 0.0546 | 0.1093 |
| P-STA | 0.1078 | 0.9375 | 0.1093 |
| P-TAL | 0.0756 | 0.3750 | 0.0546 |
| C-HUE | 0.0718 | 0.6875 | 0.0656 |
| C-APU | 0.0738 | 0.1875 | 0.0790 |
| C-NOV | 0.1093 | 0.1290 | 0.5781 |
| C-CAM | 0.0656 | 0.1056 | 0.6875 |
| C-VRA | 0.1093 | 0.2156 | 0.5781 |
Discussion
Our set of seven nuclear microsatellite markers indicates a lower level of genetic diversity as measured by allelic richness, which was corrected for variation in sample size, and observed heterozygosity in peripheral, isolated beech populations than in core populations from the continuous Carpathian range. Expected heterozygosity and inbreeding coefficient were similar (no significant differences) between populations classified as peripheral or core based on the beech distribution range in Romania. Similarity in expected heterozygosity but marked differences in observed heterozygosity and inbreeding coefficients between peripheral and core populations were reported in an American conifer species, Sitka spruce, using sequence tagged site loci ([22]). Very recently, similar levels of genetic diversity between core and peripheral populations were also reported in Scots pine using nuclear microsatellite loci ([43]). In terms of private alleles there was nearly a balance between the peripheral and core populations (6 vs. 5). However, private alleles were found in only one peripheral population (P-STA) which is located in the south-western part of the country in the proximity of the Balkan Mountains. Surprisingly, no private alleles were found in the other peripheral, isolated populations located much farther from the continuous natural distribution range than the population P-STA. Most of the private alleles detected in core populations are from the two beech populations located in Western Carpathians (Apuseni Mountains) on the other side of the Carpathian arc as the majority of the studied populations. The presence of private alleles in Apuseni Mountains might be explained by the existence of a refuge area for beech in that region ([29]). A slightly larger population differentiation was observed among peripheral populations than among core, Carpathian populations, although the latter ones are spread on a larger area and on both sides of the Carpathian mountain chain. This is consistent with the pattern observed in Sitka spruce and may be the result of more substantial gene flow among core populations than among peripheral ones or shared ancestral polymorphism ([22]). The clustering of populations and the outcomes of the Bayesian analysis partially reflect the geographic relationships between populations. Thus, the two populations from the Western Carpathians are grouped together as well as the three core populations from the other side of the Carpathian Mountains. The cluster with two peripheral populations, P-MAC and P-SNA, has strong bootstrap support (87%). These two isolated populations might have been the remnants of a migration wave coming from the Carpathian Mountains towards the Danube valley when the climate conditions were more favorable to beech. Mountain ranges were colonized first and valleys (e.g., Danube valley) were colonized rather late in the Holocene ([29]).
All sampled beech populations share the same chloroplast haplotype. A single haplotype was reported based on the same set of chloroplast microsatellite markers for the Carpathian region in a study on the entire natural distribution range of European beech ([29]). However, no sampling was done at that time in peripheral, isolated lowlands populations from south-eastern Romania. The hypothesis that one of the isolated, peripheral populations (P-MAC) located in the Macin Mountains might originate from a distinct (Balkan) glacial refugium ([10]), and not from the same putative refuge area as of the Carpathian populations (Moravian area - [29]) is not supported by our data. Numerous other haplotypes were detected based on the same set of three chloroplast microsatellites in the Balkan Mountains and in Greece, but none of these haplotypes were spread northwards ([27], [29], [34]) or observed in our sample. The fact that beech individuals from P-MAC share the same chloroplast haplotype with the Carpathian core populations indicates rather a common geographical origin (Moravian area) of the analyzed populations.
The occurrence of Fagus orientalis and F. x taurica individuals, hybrids between F. sylvatica and F. orientalis, were documented in the peripheral Macin (P-MAC) population ([12], [31]). The larger proportion of oriental-like beech individuals suggested a different evolutionary history of this remote beech population. However, first observations made on the site indicated the presence of F. sylvatica-like individuals. Moreover, typical ground flora species for beech stands in the nearby Carpathian Mountains were also identified in the beech stand from Macin ([23]). Recent statistical analyses of leaf morphology of individual trees sampled in Macin population suggested that most of the individuals are F. sylvatica-like and only a few show characters of F. orientalis ([7]).
A long distance founding event may explain the origin of the beech population (P-MAC) in Macin Mountains. This event implies the existence of bottleneck signatures which was actually found under either IAM or SMM model in population P-MAC. Moreover, evidence of a bottleneck was observed in a second peripheral population of very small size (P-SNA). The rest of the peripheral populations may also have experienced bottlenecks, but given the limited sample size of individual trees and loci used in our study, microsatellite-based bottleneck tests often have a limited power to detect recent declines of populations ([37]). An advance of beech front from the Carpathian Mountains towards the south-eastern lowlands (steppe) of Romania along river valleys or during periods of a more humid climate and long distance dissemination events is the most plausible hypothesis for the origin of the current peripheral populations. Actually, what we see at present is only a small portion of the isolated, peripheral beech stands that existed before in the region ([16], [20], [23]). A forest site with typical ground flora for beech stands but no beech individuals were identified in the Macin Mountains at the beginning of the 20th century. The lack of beech individuals on a typical site might be explained by wood extractions made by the local population ([23]). Evidence of remote populations, located several hundred kilometers away from the main species distribution range and which may originate as a result of long distance dissemination events are also found in species of related genera such as in Quercus pubescens at the northern edge of the distribution range ([6]).
In conclusion, our data suggest that the existing peripheral beech populations located at the eastern edge of the species distribution range are remnants of a wider array of small beech populations having the same geographical origin as those from the Carpathian Mountain chain. These peripheral populations are less variable than the core populations from the continuous distribution range in terms of allelic richness and observed heterozygosity. Moreover, the population differentiation among peripheral populations is higher than among core populations. This may be mainly explained by bottleneck effects in the past, of which we found evidence in two peripheral populations, and restricted gene flow with the putative origin populations from the Carpathian Mountains. The survival of these peripheral populations under extreme ecological conditions (increased temperatures, prolonged drought) makes them particularly important for research and conservation purposes.
Acknowledgements
This paper was elaborated during the COST Action FP1202 MaP-FGR. Elena Ciocîrlan was supported by the Sectoral Operational Programme Human Resources Development (SOP HRD), ID134378 financed from the European Social Fund and by the Romanian Government. We are indebted to numerous colleagues from the forest districts across the country for assisting us during the field sampling. We are grateful to an anonymous reviewer for constructive comments on a previous version of the manuscript.
References
Gscholar
Gscholar
Gscholar
Gscholar
Gscholar
CrossRef | Gscholar
Gscholar
CrossRef | Gscholar
Gscholar
Gscholar
Gscholar
CrossRef | Gscholar
Gscholar
Gscholar
Gscholar
Gscholar
Authors’ Info
Authors’ Affiliation
Nicolae Sofletea
Alexandru Lucian Curtu
Department of Forest Sciences, Transilvania University of Brasov, Sirul Beethoven-1, 500123 Brasov (Romania)
CREA SEL, Consiglio per la ricerca in agricoltura e l’analisi dell’economia agraria, Forestry Research Centre, I-52100 Arezzo (Italy)
Corresponding author
Paper Info
Citation
Ciocîrlan E, Sofletea N, Ducci F, Curtu AL (2017). Patterns of genetic diversity in European beech (Fagus sylvatica L.) at the eastern margins of its distribution range. iForest 10: 916-922. - doi: 10.3832/ifor2446-010
Academic Editor
Fulvio Ducci
Paper history
Received: Mar 29, 2017
Accepted: Aug 23, 2017
First online: Dec 10, 2017
Publication Date: Dec 31, 2017
Publication Time: 3.63 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2017
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 53857
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 43923
Abstract Page Views: 4152
PDF Downloads: 4451
Citation/Reference Downloads: 23
XML Downloads: 1308
Web Metrics
Days since publication: 3164
Overall contacts: 53857
Avg. contacts per week: 119.15
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Jul 2026)
Total number of cites (since 2017): 12
Average cites per year: 1.20
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Genetic diversity of core vs. peripheral Norway spruce native populations at a local scale in Slovenia
vol. 11, pp. 104-110 (online: 31 January 2018)
Technical Reports
Conservation and use of elm genetic resources in France: results and perspectives
vol. 13, pp. 41-47 (online: 03 February 2020)
Research Articles
Clonal structure and high genetic diversity at peripheral populations of Sorbus torminalis (L.) Crantz.
vol. 9, pp. 892-900 (online: 29 May 2016)
Editorials
Workshop COST E52 “Evaluation of beech genetic resources for sustainable forestry”
vol. 2, pp. 104 (online: 10 June 2009)
Commentaries & Perspectives
The genetic consequences of habitat fragmentation: the case of forests
vol. 2, pp. 75-76 (online: 10 June 2009)
Research Articles
Identification and molecular characterization of LTR and LINE retrotransposable elements in Fagus sylvatica L.
vol. 2, pp. 119-126 (online: 10 June 2009)
Research Articles
Comparison of genetic parameters between optimal and marginal populations of oriental sweet gum on adaptive traits
vol. 11, pp. 510-516 (online: 18 July 2018)
Review Papers
Genetic diversity and forest reproductive material - from seed source selection to planting
vol. 9, pp. 801-812 (online: 13 June 2016)
Research Articles
Networking sampling of Araucaria araucana (Mol.) K. Koch in Chile and the bordering zone of Argentina: implications for the genetic resources and the sustainable management
vol. 2, pp. 207-212 (online: 22 December 2009)
Research Articles
Weak isolation by distance and geographic diversity gradients persist in Scottish relict pine forest
vol. 11, pp. 449-458 (online: 02 July 2018)
iForest Database Search
Search By Author
Search By Keyword
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords
PubMed Search
Search By Author
Search By Keyword







