Towards an optimal sampling effort for paternity analysis in forest trees: what do the raw numbers tell us?
iForest - Biogeosciences and Forestry, Volume 5, Issue 1, Pages 18-25 (2012)
doi: https://doi.org/10.3832/ifor0606-009
Published: Feb 27, 2012 - Copyright © 2012 SISEF
Short Communications
Abstract
The ever-increasing use of paternity analysis to estimate the dispersal capability of forest trees calls for a quantitative evaluation of potential errors due to sampling design. Previous studies on optimal sampling strategies for seed trapping experiments suggested a link between sampling effort and error rate in the reconstruction of the seed dispersal kernel. We considered 92 papers on paternity analysis to quantitatively assess the sampling strategy used to study the characteristics of pollen dispersal patterns (pollen immigration rate, distribution of male reproductive success and estimates of pollen dispersal kernel parameters). For each studied stand we report data on the sampling effort (the total number of sampled seeds, the number of mother trees and the number of seeds per mother tree) and additional information on the studied species and characteristics of the sampling areas. The reviewed papers used a median of 8 mother trees (acting as pollen traps in paternity analysis studies), a median of 29 seeds per mother tree and a median of 240 total sampled seeds. These are values (especially the number of mother trees) lower than usually found in classical seed trapping studies, for which accuracy and precision of seed dispersal estimates had already been assessed. These findings underline the need of evaluating the consequences of realistic sampling efforts on the estimation of parameters describing the pollen dispersal pattern to provide the basis for meaningful guidelines to paternity analysis.
Keywords
Sampling scheme, Pollen-mediated gene flow, Dispersal, Seed trapping, Inverse modeling
Introduction
Paternity analysis is based on the evaluation of the compatibility between the genotypes of adult male individuals and the male genotypic contribution to seeds, and is the preferred method for estimating pollen-mediated gene flow in natural plant populations. Tracking of pollen movements that led to successful fertilization of ovules provides an estimate of the “realized” gene flow. Paternity analysis requires the sampling of all males (hereafter referred to as “potential pollen donors”) within a defined area and a sample of seeds collected from fruiting trees acting as pollen traps (hereafter “mother trees”) from the same area. In monoecious species, mother trees are also potential pollen donors. After genotyping all potential pollen donors, mother trees and collected seeds, the assignment of paternity can be carried out with various analytical methods whose pros and cons have recently been reviewed by Jones et al. ([58]). The main aim of paternity analysis is to correctly identify the true father of any collected seed (or to detect immigrant pollen when no local pollen donor is compatible with seed genotype). Results from paternity analysis (pollen immigration rate, distribution of male reproductive success and estimates of pollen dispersal kernel parameters) are thought to be affected by the resolution of the marker set used and by genotyping errors ([16], [5]).
Paternity analysis is a powerful tool for the study of within-population pollen dispersal patterns. The short distance component of the dispersal pattern has strong influence in shaping fine-scale genetic structure, that in turn determines the rate and direction of microevolutionary changes at the population level ([79]). In isolated and low density populations, paternity analysis allows to trace pollination events at a larger scale (the long distance component of the dispersal pattern). It is well-established that forest tree pollen is able to travel hundreds of kilometres and evidence is accumulating that after such long-distance dispersal events pollen is viable and can successfully fertilize seeds ([110], [87], [17]). The possibility of quantifying effective pollination over long distance has profound consequences on the study of how genes have travelled in past and ongoing tree migrations, and may contribute to a sound forecasting of tree responses to anthropogenic and natural global changes ([89]). In addition, risk assessment of pollen escape from GM plantation and predictions on the spread of invasive alien species strongly depend on an accurate estimate of the long distance component of pollen dispersal kernel ([109]).
The impact of sampling scheme on the results from paternity analysis has received relatively little attention. A few studies assessed the effects of location and number of seed traps and number of seeds collected in each trap in classical seed trapping experiments. Data from these studies are usually analyzed following a backward approach (so-called “inverse modeling”) aimed at reconstructing the dispersal kernel from the spatial location and the fecundity of potential parents and the spatial pattern of seeds collected from traps ([82]). Skarpaas et al. ([95]) used a simulation approach to optimize seed trap sampling design around a point source. They showed how traps arranged in transects and sectors provide a better kernel estimation than other sampling schemes. Annuli and grid arrays outcompeted other schemes only when the anisotropy of dispersal was unknown. Pielaat et al. ([76]) found that a trade-off between nearby and distant sampling is needed to accurately characterize the tail of the dispersal kernel. It is also well known that a random placement of traps within a rectangular or circular area determines an uneven sampling of distance classes, leading to the over-representation of the intermediated ones ([42]).
So far, the most relevant study whose results on sampling effort can be easily extended to the case of paternity analysis is the one by Robledo-Arnuncio & Garcia ([85]). They proposed a maximum-likelihood procedure to estimate the seed dispersal kernel from the exact identification of seed sources, as in the case of parentage assignment based on genetic compatibility. This method (Competing Sources Model - CSM) works out the problem of the uneven distribution of mother-trap potential distances by taking into account the spatial arrangement of seed traps relative to source plants. It provides better estimates of seed dispersal kernel parameters compared to standard maximum likelihood fitting used in inverse modeling ([85]). Jones & Muller-Landau ([56]) compared different approaches to estimate dispersal kernel parameters and confirmed that CSM is among the most accurate and robust methods. From the results of simulations on different sampling scenarios in Robledo-Arnuncio & Garcia ([85]) some practical recommendations on sampling effort emerged. For example, it was shown that fewer seeds are required to properly estimate the average dispersal distance (hereafter “δ”) with respect to the shape parameter (hereafter “b”) of the exponential power kernel (a widely used and flexible curve to describe seed and pollen dispersal), and that for a fixed total number of seeds increasing the number of traps is more useful than collecting more seeds per trap for reducing estimation uncertainty.
Sampling for paternity analysis differs substantially in the number of traps from the one for inverse modeling. In paternity analysis a large number of mother trees per population is rarely sampled (see for instance [91] and [72] for some exceptions), whereas in inverse modeling 10 to 300 traps are usually placed ([99], [73], [57]). However, quantitative assessments or qualitative indications about the spatial arrangement of mother trees and the total number of seeds, mother trees, and seeds per mother tree can seldom be found in the literature on paternity analysis. An inadequate or insufficient sampling effort (too few sampled seeds and/or mother trees) can lead to a biased estimation of the within-population pollination pattern. In addition, the lack of standard sampling methods limits the comparison among studies to draw general conclusions.
In the present work we review 92 paternity analysis papers to provide a quantitative assessment of the sampling strategy upon which the estimate of pollen-mediated gene flow rate, the reconstruction of pollen dispersal kernel and the description of male reproductive success distribution are based. We report data on the sampling effort (the total number of sampled seeds, the number of mother trees and the number of seeds per mother tree) and discuss possible consequences and limitations in the sampling scheme on paternity analysis results.
Materials and methods
We searched for published studies using paternity analysis to estimate pollen-mediated gene flow in forest trees. We used 3 different databases (Google ScholarTM, ISI Web of ScienceTM and ScopusTM) for surveying the literature. The key-words used were: paternity analysis, tree*, pollen, genetic* and gene flow. We also tracked references: (i) within the articles found; (ii) from review papers on gene flow in forest trees ([15], [3]); (iii) from Table 4 in Bittencourt & Sebbenn ([7]); and (iv) from Appendix A-1 in Wang et al. ([106]). Studies based on both mating models (as MLTR - [83]) and pollen pool heterogeneity (as KINDIST - [86]) were excluded since they require only to genotype seeds (and mother trees). Studies based on methods such as MLTR ([83]) and KINDIST ([86]) were excluded since they require only to genotype seeds and mother trees. Moreover, we did not consider a few studies for which we were unable to obtain the full-text. Since the focus of our paper is on the sampling strategy, studies based on previously published data or metanalyses were also excluded in order to avoid duplicates. We included paternity studies carried out in seed orchards and studies investigating gene flow among closely related species (e.g., Quercus spp.).
For each selected paper we recorded:
- information on sampling strategy: the total number of sampled seeds, the number of mother trees and the average number of seeds per mother tree;
- characteristics of the studied population: number of potential pollen donors, number of potential pollen traps (female individuals in the population for dioecious species), area, tree density;
- the studied species, its family and taxonomic group, breeding system and primary pollination vector;
- the method and molecular markers used for paternity assignment.
In monoecious species the number of male individuals is equal to the number of female individuals. Whenever life history traits could not be found in the text, we tried to gather them from other sources (e.g., on-line databases), or in some cases from personal communications with the Authors. When density was not available in the text it was inferred dividing the number of individuals within the study population by the stand area. As a general rule, papers either providing a poor description of the sampling design or lacking many essential data were excluded from our dataset.
In papers with multiple stands (e.g., when a system of several forest fragments is studied), data were collected for every stand where at least one seed was genotyped to estimate the pollen-mediated gene flow characterizing that stand (or a group to which the specific stand belongs as, for instance, in [65]). In studies where the same stand was analyzed in two or more consecutive years, if data on sampling effort were reported for each year and they differed, we considered each year as an independent data point.
As for studies carried out in seed orchards or for species that reproduce vegetatively, the number of ramets was taken as the number of individuals, since ramets (rather than genets) represent spatially distinct pollen sources.
Results and discussion
General contents and sources
We collected data from 92 papers published from 1992 to September 2011. Among them, 14 were also present in Burczyk et al. ([15]) and 27 in Ashley ([3]). In the latter work, the literature search was only on native plants (cultivated trees and crops were excluded) and microsatellite-based studies, but experiments based on parentage analysis were also taken into account. The author stated that her search was not exhaustive, but “broadly inclusive and representative”.
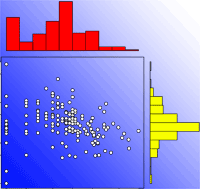
The number of papers published per year increased until 2008, followed by a slight decrease in the following years. The general growing trend seems related to the increasing availability of microsatellite markers and to the development of methods based on maximum likelihood assignment of paternities (Fig. 1). The most used methods (simple exclusion - SE, neighborhood model - NM, and maximum likelihood - ML) have similar sampling requirements. The main difference concerns the spatial distribution of pollen donors to be sampled. As for SE and ML-based studies, all potential pollen donors within the sampling area must be sampled, whereas for NB-based studies sampling is carried out within circular areas of a given radius surrounding n mother trees.
Fig. 1 - Number of published studies per year. Different colors represent: (a) different methods in the analysis of paternity data; and (b) different molecular markers. In papers based on maximum likelihood methods, data analysis was usually performed by the CERVUS ([59]) and FaMoz ([41]) software. In papers based on the neighbourhood model, data analysis was usually performed according to the methods presented in Burczyk et al. ([13]) - implemented in the NM+ program by Chybicki & Burczyk ([23]) - and Oddou-Muratorio et al. ([72]).
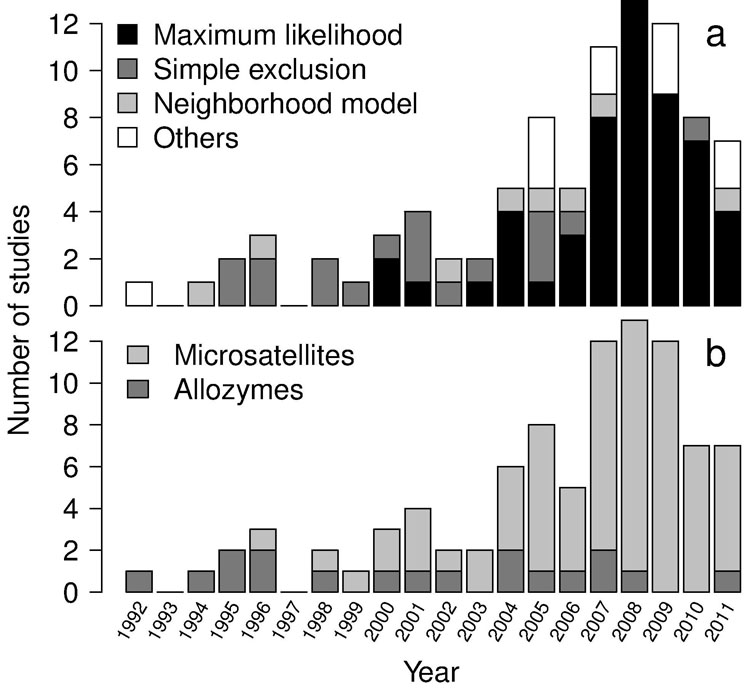
Few studies performed paternity analysis using multiple analytical methods (n=8, 9%). The use of multiple approaches for the estimation of gene flow rates has been recently proposed to overcome possible drawbacks related to specific methods and/or weaknesses due to low-resolution marker sets ([5], [58]). Almost half the papers (47%) were published in only 4 journals, with Heredity and Molecular Ecology clearly representing the preferred journals for gene flow studies based on paternity analysis (respectively, 15% and 14%), followed by Forest Ecology and Management and Conservation Genetics (Fig. 2).
Fig. 2 - Number of studies published per journal. Journals with only 1 published study were excluded.
Studied species and sampling areas
The collected papers covered a total of 81 different species. Most studies (n=75, 81%) were conducted on Angiosperms, whereas only 17 studies focused on Gymnosperms. Fagaceae (16) and Pinaceae (12) were the most represented families and, together with Fabaceae and Dipterocarpaceae, represented the studied species in almost half of the collected papers. Overall, species from 30 different families were studied. The large number of gene flow papers on Fagaceae was also reported by Ashley ([3]). With regard to the primary pollination vector, there were 46 studies on insect-pollinated species (50%), 38 on wind-pollinated species (41%), while in the remaining 8 articles the studied species relies on mammals, birds, or multiple vectors for pollination.
We obtained 187 data points from the 92 collected papers (see Appendix 1). Fifty-eight papers (63%) investigated a single stand, providing a single data point. Papers with more than one data point were fairly frequent (n=34, 37%). In general, this is due to the analysis of multiple stands within the same article. Wang et al. ([107]) studied pollen-mediated gene flow in 28 fragments of Pinus tabulaeformis in an urban landscape. According to our criteria, we retained 25 data points, the largest number of data points from a single study in our dataset. Also Lander et al. ([65]) sampled a large number of forest fragments to estimate pollen immigration, but only a half of them matched our criteria, providing 13 data points. On the other hand, 11 papers investigated gene flow in the same stand over multiple years (usually, 2 or 3 consecutive years). Irwin et al. ([53]) highlighted that single-season studies may not capture temporal variability in pollen exchange, especially in perennial plants where flowering does not occur every year. Therefore, multi-year analyses were advocated for obtaining accurate estimates of pollen-mediated gene flow patterns.
In general, our knowledge on pollen-mediated gene flow for a species is based on a single study. Only 14 species were analyzed in more than one paper. Quercus robur was investigated in three papers, while Araucaria angustifolia, Cryptomeria japonica, Eucalyptus grandis, Eurycorymbus cavaleriei, Fagus sylvatica, Picea abies, Populus nigra, Prunus avium, Pseudotsuga menziesii, Q. macrocarpa, Q. salicina, Shorea leprosula and Sorbus torminalis were studied twice. The lack of independent estimates, along with the usually low degree of comparability among pollen-mediated gene flow studies, makes generalization on single estimates far-fetched. Studies designed for allowing meaningful comparisons among gene flow rates estimated in different ecological conditions are also rare. The importance of such comparative studies in characterizing the pollen dispersal capability of a species is discussed in Piotti et al. (in press).
The median area of the 118 stands for which we found sufficient information was 7.42 ha. Small stands (≤ 1 ha) are fairly common (24%). Two thirds were smaller than 20 ha, whereas larger stands (≥ 100 ha) are rare (8%). This pattern is likely determined by the rapid increase in sampling and genotyping effort with size of the sampled area. Studies on large areas are suitable for the detection of rare long-distance dispersal events, though they are feasible only when species are present at low density. However, Hardy ([49]) pointed out that, despite their great importance, the measures of dispersal obtained in such studies might not be representative of species with similar pollination syndrome at higher densities. Comparying two natural Populus trichocarpa stands dramatically different as for density (993 vs. 0.2 males km-2) and area (19.6 vs. 31400 ha), Slavov et al. ([97]) found large differences in pollination patterns, although the authors themselves warned about the difficulty of comparing such different areas. Piotti et al. (in press) compared two close Fagus sylvatica stands with regular densities characterized by different management regimes. They found a more skewed pollen dispersal distance distribution in the managed area whose density is about one-third of the unmanaged one (57 vs. 163 trees ha-1). Information on the size of sampling areas can also be useful to understand if limits in sampling scale may downwardly bias dispersal range estimates. Nonetheless, heterogeneity of experimental setups and methods used, as well as variation in population densities, pollination syndromes, pollen terminal velocity, stand isolation, etc. should be carefully taken into account in data collection and analysis for future works on this topic. However, methods to estimate dispersal parameters taking advantage of spatially censored data ([57]) or assuming the immigration rate to be a function of dispersal kernel ([43]) are already available but rarely applied. These approaches allow to take into account immigration events of unknown origin in the estimation of the dispersal curve, usually resulting in a substantially higher mean dispersal distance ([78], [24]).
Sampling strategy
In any of the papers collected for this study, we found neither exhaustive justifications for the sampling strategy adopted, nor references to any guideline for sampling strategy. An exception was the paper by Oddou-Muratorio et al. ([72]), who stated that “the objectives for both years were to sample all possible distances between mother trees, and to maximize the number of mother trees in the middle part of the study site”. A paragraph in their discussion focused on the methodological insights for the estimation of the dispersal kernel. Many papers reported a map of the sampling area showing the location of adult individuals with mother trees indicated by different symbols (e.g., [71], [70], [28]). Based on visual inspection of these maps, no clear patterns in the choice of mother trees can be recognized. Their location varied from clustered in the centre of the stand to scattered throughout the entire sampling area. We did not find any statement about a random choice of mother trees in the collected papers. Given this lack of indications and our experience on the difficulties in sampling seeds from forest trees, we feel that such diverse sampling schemes might arise from practical constraints rather than a thorough a priori evaluation. As pointed out in methodological papers on seed dispersal modeling, intuitively designed experiments are likely to lead to incomparable and non-representative results ([111], [99]).
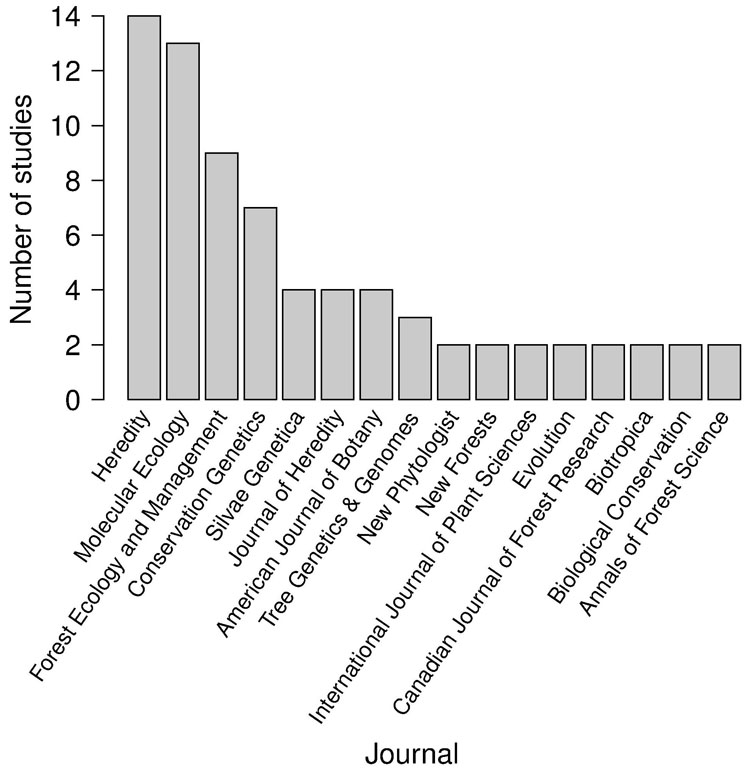
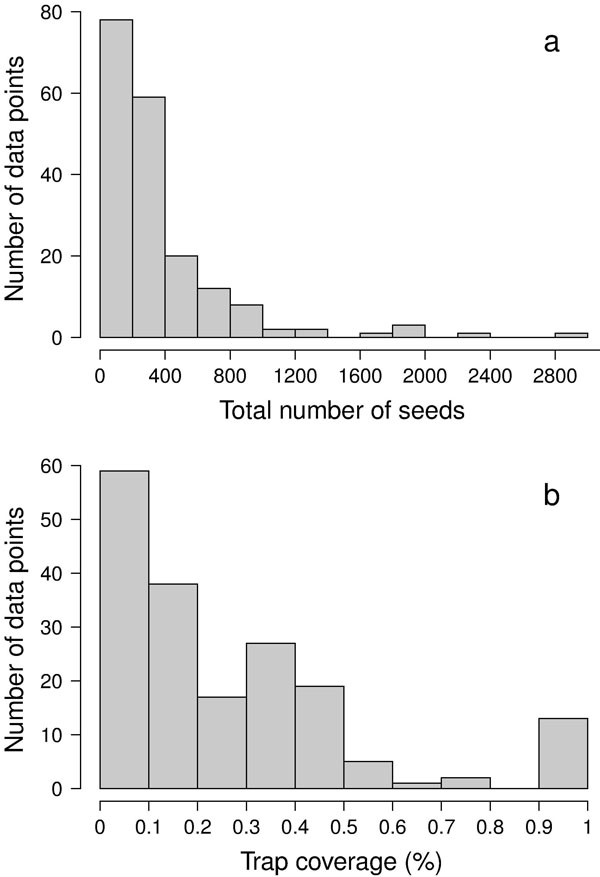
Besides the spatial positioning of mother trees, the other crucial factor in designing a solid sampling scheme for paternity analysis is the sampling effort, represented by the total number of sampled seeds (i.e., number of mother trees x number of seeds per mother tree). In the collected papers, we found a median number of 8 mother trees (mean = 11.3 ± 13.6 SD - Fig. 3), 29 seeds per mother tree (mean = 45.6 ± 74.7 SD - Fig. 3) and 240 total sampled seeds (mean = 356.1 ± 423.1 SD - Fig. 4a). These values are lower than those usually found in classical seed trapping studies. This difference is likely to depend on the need for larger samples to infer dispersal kernel’s parameters without genetic data and, on the other hand, on the significant investment of time and resources to collect genetic data ([56]). As a measure of the coverage of potential pollen traps in a stand (trap coverage) the ratio of mother trees over the total number of female trees was calculated. The median value of trap coverage was 0.18 (mean = 0.28 ± 0.27 SD - Fig. 4b). The above parameter was not correlated with tree density, but it negatively depends on the total number of female trees, indicating that trap coverage is more exhaustive in small populations. The low trap coverage in large stand studies can be related to practical and economic limitations. Nevertheless, a sufficiently high trap coverage is desirable to decrease confidence interval of parameter estimates and increase comparability among results from different experiments.
Fig. 3 - Distributions of the number of mother trees (x-axis) and number of sampled seeds per mother tree (y-axis) in log scale. White dots represent the 187 data points collected for this study.
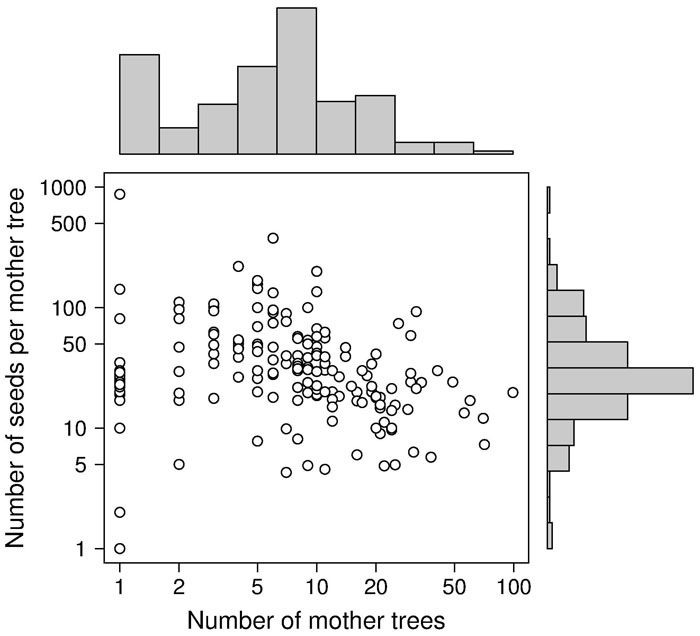
Fig. 4 - Distributions of (a) the total number of seeds, and (b) the percentage of trap coverage characterizing the 187 data points.
Our data on the distribution of sampling effort in published paternity analyzes are comparable with the ones from Robledo-Arnuncio & Garcia ([85]), though their work was focused on studying seed dispersal with seed trapping and genetic data. By using a simulation approach, they tested the performance of the CSM by randomly placing 20, 100 and 200 traps in a squared area, and sampling 1 to 50 seeds per trap (total number of sampled seeds: 200-1000). They found that the CSM performs well in the estimation of the δ parameter even for a relatively small number of seeds (200), whereas ≥ 500 seeds are needed to obtain an accurate estimate of the b parameter of the dispersal kernel. As the authors noted, the b parameter is more sensitive than the δ parameter to decreasing number of total seeds and seed traps. The minimum number of traps they simulated was 20, that is low for classical seed trapping experiments (being therefore adequate for the aims of their paper), but almost double the mean number of mother trees in paternity studies. Among the few indications in the literature about sampling effort in paternity analysis, Oddou-Muratorio et al. ([72]) noted that increasing the number of mother trees from 14 to 60 (in the same area in two consecutive years) sensibly reduces the confidence interval of the parameters of the dispersal kernel. On the other hand, the authors pointed out that sampling a high number of seeds per mother tree, that usually limits the number of mother trees, could be more adequate for the estimation of individual selfing rates. This implies that sampling strategy in paternity analysis should be fine-tuned to meet the specific aims of an experiment.
Conclusions
Although some results on the consequences of different sampling schemes are available for seed trapping studies (with or without genetic assignment of seeds), the case of paternity analysis, usually based on a lower sampling effort, is poorly investigated. Our data collection from the literature on paternity analysis in forest trees showed a potential lack of knowledge about the effects of low numbers of mother trees, seeds per mother tree and total sampled seeds on estimates usually obtained to describe within-population pollination patterns: (i) pollen immigration; (ii) male reproductive success; and (iii) parameters of the pollen dispersal kernel. Only in 29 out of the 187 collected data points (15%) the number of mother trees is higher than 20, the lowest number of traps taken into account by Robledo-Arnuncio & Garcia ([85]). This means that for 85% of our collected data points we have little idea about how accurate and precise the estimates from paternity analysis can be. From Table 2 in Robledo-Arnuncio & Garcia ([85]) we know that the relative root mean square error (RMSE - a measurement of both accuracy and precision of an estimate normalized to the expected value) is ~0.04 for the δ parameter and ~0.10 for the b parameter when 500 seeds were sampled. RMSEs increased to ~0. 07 and ~0.17, respectively, when the total number of seeds decreased to 200. Errors roughly increase with the inverse of the square root of the total number of seeds sampled, as expected from classical statistical theory. Consequently, when the sampling effort is scarce non negligible errors in estimates are expected, in particular for the b parameter. Leonarduzzi et al. (in preparation), relying on the distribution of sampling effort presented here, explore the consequences of realistic sampling strategies on the reconstruction of different dispersal kernels to provide the basis for meaningful guidelines for paternity analysis.
Acknowledgments
The authors are grateful to Elia Vajana for help during literature survey and data collection and to Brad Oberle for English revision and suggestions on an early version of the manuscript. We also thank two anonymous reviewers for helpful comments.
References
Gscholar
Gscholar
CrossRef | Gscholar
Gscholar
Gscholar
Gscholar
CrossRef | Gscholar
CrossRef | Gscholar
Gscholar
Gscholar
Gscholar
Authors’ Info
Authors’ Affiliation
S Leonardi
P Menozzi
A Piotti
Department of Environmental Sciences, University of Parma, v.le G.P. Usberti 11/A, I-43124, Parma (Italy)
Corresponding author
Paper Info
Citation
Leonarduzzi C, Leonardi S, Menozzi P, Piotti A (2012). Towards an optimal sampling effort for paternity analysis in forest trees: what do the raw numbers tell us?. iForest 5: 18-25. - doi: 10.3832/ifor0606-009
Academic Editor
Marco Borghetti
Paper history
Received: Dec 06, 2011
Accepted: Feb 01, 2012
First online: Feb 27, 2012
Publication Date: Feb 27, 2012
Publication Time: 0.87 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2012
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 62121
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 50715
Abstract Page Views: 4306
PDF Downloads: 5552
Citation/Reference Downloads: 38
XML Downloads: 1510
Web Metrics
Days since publication: 5238
Overall contacts: 62121
Avg. contacts per week: 83.02
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Mar 2025)
Total number of cites (since 2012): 12
Average cites per year: 0.86
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Weak isolation by distance and geographic diversity gradients persist in Scottish relict pine forest
vol. 11, pp. 449-458 (online: 02 July 2018)
Commentaries & Perspectives
The genetic consequences of habitat fragmentation: the case of forests
vol. 2, pp. 75-76 (online: 10 June 2009)
Research Articles
Seed trait and rodent species determine seed dispersal and predation: evidences from semi-natural enclosures
vol. 8, pp. 207-213 (online: 28 August 2014)
Technical Reports
Conservation and use of elm genetic resources in France: results and perspectives
vol. 13, pp. 41-47 (online: 03 February 2020)
Technical Advances
Gene flow in poplar - experiments, analysis and modeling to prevent transgene outcrossing
vol. 5, pp. 147-152 (online: 13 June 2012)
Research Articles
Pollen contamination and mating patterns in a Prosopis alba clonal orchard: impact on seed orchards establishment
vol. 12, pp. 330-337 (online: 14 June 2019)
Research Articles
Delineation of seed collection zones based on environmental and genetic characteristics for Quercus suber L. in Sardinia, Italy
vol. 11, pp. 651-659 (online: 04 October 2018)
Short Communications
Evidence of Alectoris chukar (Aves, Galliformes) as seed dispersal and germinating agent for Pistacia khinjuk in Balochistan, Pakistan
vol. 14, pp. 378-382 (online: 22 August 2021)
Research Articles
Networking sampling of Araucaria araucana (Mol.) K. Koch in Chile and the bordering zone of Argentina: implications for the genetic resources and the sustainable management
vol. 2, pp. 207-212 (online: 22 December 2009)
Research Articles
Seed germination traits of Pinus heldreichii in two Greek populations and implications for conservation
vol. 15, pp. 331-338 (online: 24 August 2022)
iForest Database Search
Search By Author
Search By Keyword
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords
PubMed Search
Search By Author
Search By Keyword






