Gene flow in poplar - experiments, analysis and modeling to prevent transgene outcrossing
iForest - Biogeosciences and Forestry, Volume 5, Issue 3, Pages 147-152 (2012)
doi: https://doi.org/10.3832/ifor0618-005
Published: Jun 13, 2012 - Copyright © 2012 SISEF
Technical Advances
Collection/Special Issue: COST Action FP0905
Biosafety of forest transgenic trees and EU policy directives
Guest Editors: Cristina Vettori, Matthias Fladung
Abstract
The demand for energy and forestry products is globally increasing, raising the question if traditional breeding programs are efficient and fast enough to keep up with these demands. A possible solution seems to be the use of genetic engineering techniques, since classical breeding strategies are time-consuming and limited by species barriers. Besides the advantages of genetic engineering technologies, concerns are also raised by scientists regarding these methods. Consequently, risk analysis of genetic modified trees in plantation forestry is a fundamental research topic. This paper presents a sequence of steps in risk analysis dealing with genetic modified poplar clones in the natural environment, ranging from investigations of flowering phenology, to molecular identification of gene flow patterns and their statistical interpretation, to modeling approaches to simulate different scenarios of plantations using genetic modified poplars in realistic European landscapes. All steps are evaluated for their potential to forecast the risk of outcrossing of gene constructs into native populations. The application of the results achieved to short rotation plantations are discussed.
Keywords
Poplar, Genetic Modified Trees, Flowering Phenology, Germination, Simulation Models
Introduction
Traditional forest breeding programs have been initiated in the middle of the last century in order to improve growth rate, stem form and wood quality. Attempts have been also made to improve disease resistance ([8]). In these programs two main strategies to propagate desired material were applied. The first strategy involves seed harvesting from clonal seed orchards and selection according to their performance under specific conditions. The other method uses vegetative propagation of superior trees for direct use in plantations. Both methods are time-demanding, since selection of the appropriate trees can only take place at the age of rotation ([23]). So far, traditional forest breeding programs have only undergone 3-4 cycles.
Because of the world-wide high demand for forestry products and renewable energy, traditional forest breeding programs do not seem efficient enough to keep up with these demands. In this context, the use of more advanced techniques such as genetic engineering may be necessary to produce new varieties of trees for plantation forestry with high efficiency and within a short time span. In particular, long life cycle and extended vegetation phase are major shortcomings limiting the efficiency of traditional breeding programs ([2]). In this respect, genetic engineering is the only way (so far) escaping this deficit and realizing the desired traits in the target species within a reasonable time frame.
Besides the advantages of increasing the speed of breed selection, genetic engineering allows to circumvent natural selection. Genetic modified organisms released into the natural environment are not tested for their effects on the habitat nor for their selective value in competitive situations. Furthermore, genetic engineering does not only speed up the breeding process, but also override the incompatibility barriers between species, in that genes from different taxa may become incorporated into a single genome ([2]). For these reasons, it is generally recommended by the scientific community to prevent these artificial gene combinations to escape their hosts and outcross into native populations ([6]).
In commercial plantations fast-growing species of the genus Populus (Salicaceae) are already intensively cultivated ([14]) and their hybrids widely used in breeding programs ([10], [32]). In addition, the genome of one poplar species has been already fully sequenced ([34]), which is the prerequisite for a wide range of genetic modification ([5]). Traits of interest, like accelerated growth, wood properties and stress tolerance, are mainly quantitative traits controlled by a large number of genes. In such cases the modification of a single gene would be not sufficient and larger constructs are needed ([2]), giving even more opportunities for mixing genomic informations from different taxa.
Evidence on transgene constructs originated from transgenic crops and found in wild populations has been already reported ([3]). Against this background, several aspect of biosafety are discussed in literature concerning the usage of genetically modified trees in commercial plantations ([15]). This includes outcrossing, gene transfer between non-native/bred and native tree species, as well as transgene instability, to name just a few. In order to prevent outcrossing of these gene constructs by pollen or seeds, researchers are additionally investigating traits controlling flowering and sterility ([16]). So far, transgenic poplars carrying insect resistance genes are only commercialized in China ([13]). In Europe no such plantations do exist and therefore other methods are needed for a sound evaluation of the impact of genetically modified poplars on native European Black poplar (Populus nigra L.) populations.
This paper summarizes the main results from three research projects dealing with different aspects of gene flow and transgene outcrossing between poplar taxa in Germany. The basic concept of all the projects was the analysis of gene flow between native Black poplar trees (Populus nigra L.) and its commercial hybrid (Populus × canadensis Moench). In this scenarios, P. × canadensis is regarded at as the genetic modified taxon. P. × canadensis is carrying species-specific genes from eastern cottonwood (Populus deltoides Bartr. ex. Marsh.), which have been used as indicator genes for transgene outcrossing.
The projects were focused on the whole chain of transgene outcrossing and establishment in the wild. The investigations carried out did range from pollen flow and flowering to seed dispersal and germination. Additionally, modeling techniques have been used to make predictions and to analyze scenarios related to the use of genetic modified trees in real landscapes. Next chapters will cover some of the aspects considered by the above-mentioned projects.
Indication of potential gene flow derived by flowering phenology
The first step for assessing the outcrossing potential of genetic modified trees after their introduction into the landscape is monitoring their flowering phenology. Only in the case of a certain overlap of flowering between bred and native species, these two taxa may interbred. Genetic modification of trees will not alter timing and duration of flowering, although efforts are undergoing to completely prevent flowering of transgenic trees ([16]). As an alternative, trees may be modified in such a way to ensure no overlap in flowering time with their native relatives. Because the study system of this paper involves a hybrid between the European P. nigra and the American P. deltoides, flowering time may have changed reducing the risk of outcrossing to a minimum.
In this study, the flowering phenology of P. nigra and P. × canadensis was monitored at two different locations in central Germany, where both taxa were present in close vicinity ([25]). Observations were carried out in spring 2006, after a very long winter followed by a rapid increase of temperatures. As a consequence, both taxa did flower at the same time, creating the conditions for outcrossing. Interestingly, males did flower (on average) before their female counterparts, irrespective of the taxa (Fig. 1). Other studies on flowering phenology in poplars from Belgium over two consecutive years (1999 and 2000) did also reveal a certain overlap in flowering between taxa ([35]), as well as a high level of variation between both years and taxa. In the year 2000 P. × canadensis bloom occurred slightly earlier than P. nigra in Belgium. Conversely, data collected in the same year from the nature protection area “Kühkopf-Knoblochsaue” in Germany showed a time gap in flowering between taxa large enough to keep their pollen clouds separated (Gebhardt, unpublished).
Fig. 1 - Flowering time span of P. nigra and P. × canadensis in 2006 at the Nature Protection Area “Ederaue bei Cappel und Obermöllrich” in central Germany. Both taxa did flower at the same time, with male flowers occurring before female catkins.
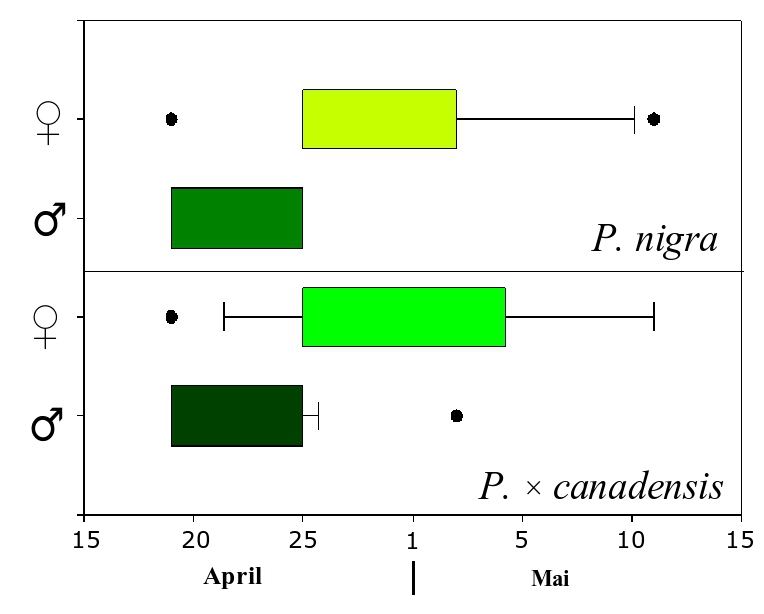
Although only a limited amount of observations are available, a first conclusion will be that hybrids flower generally earlier than the native P. nigra and a second conclusion is that males do flower before the female catkins are fully susceptible in both taxa accordingly. The above pattern results in a higher probability of female hybrids to be pollinated by male P. nigra. Our findings raises several questions about the importance of climatic conditions and the frequency of extreme events, like the long winter in 2006, for the maintenance of diversity in poplars. The answers to these questions may also be important for other pioneer species. For the maintenance of genetic diversity it would be very interesting to know if the sequence of flowering within one species does change with climatic conditions. In the case that the sequence would stay constant, in most years only a subset of all trees in the population would mate and the resulting seeds would carry only a part of the genetic diversity available at the whole population. More investigations are needed on this topic.
Gene flow assessment by molecular techniques
Mating scenarios are difficult to be identified by looking only at the flowering phenology of the studied trees. In particular, in the case of large populations spatially spread along rivers and characterized by pollen and seed dispersal over long distances, a pure observational approach is not sufficient. Molecular methods have been used to identify parents and their corresponding offspring. By the use of a set of microsatellite markers ([9]) and the computer software “CERVUS” ([22]) one may infer gene flow from genetic patterns. In order to investigate gene flow in a natural European landscape it is necessary to unambiguously distinguish the pure species from the many different hybrid clones widely used in plantations. Hybrid clones are all very similar in their genetic composition, being descendants from only a few mating parents. An efficient method to identify these clones with minimal efforts was developed by Rathmacher et al. ([30]), based a small set of seven microsatellite markers.
The above marker set has been used to characterise 566 adult trees of both taxa as well as 380 individuals from a natural regeneration site within a nature protection area in central Germany ([29]). Some 3000 seeds have been harvested directly from 12 specific mother trees (six for each taxon) and 2606 seeds genotyped using the mentioned marker set. Hence, the maternal genetic contribution of each seed could be unambiguously be identified. Paternity analysis allowed to identify the true father for most of the seeds analyzed. Because all adult trees in the stand were mapped, the reproductive effective pollen dispersal distance (i.e., the distances a pollen grain did travel to pollinate a flower on the mother tree) was calculated for each seed. Our results indicate that most of the effective pollinations or matings (75%) occurred within a distance < 1000 m. Only a very limited proportion of effective pollinations occurs at distances > 2 km. Tabbener & Cottrell ([33]) for P. nigra found pollination distances between 25-350 m and DiFazio et al. ([12]) reported mean pollination distances for Populus trichocarpa between 140-1100 m, with a strong dependency on the area sampled. This finding have been confirmed by Pospísková & Sálková ([28]), who found effective pollination distances of 10-230 m within a population of 65 adult trees and 30 analyzed seedlings of P. nigra along the Morava river.
Similar patterns are observed for seed dispersal. We performed a parentage analysis on seedlings from the natural regeneration area, and both parents were successfully identified for 169 seedlings (44.5% of the total - [29]). In the case where sex was unknown, we applied a very conservative assumption defining the closer parental tree as mother tree. Rathmacher et al. ([29]) revealed seed dispersal range of 23-7000 m. However, the main part of seeds (86%) traveled less than 1000 m and only a very small amount (1%) have been traveling more than 2 km. Because of the high exclusion power needed for parentage analysis ([22]), studies on seed dispersal in literature are few. There is only one other study on explicit seed dispersal estimation for P. nigra ([28]). Within a small population along the Morava river, they found seed dispersal distances ranging from 163 to 370 m. The difference in respect to our results may be due to the small scale of their investigation.
Nevertheless, these results have to be interpreted with caution. Only effective pollen and seed dispersal distances have been measured, and the true pollen cloud or seed availability at a specific site may be completely different. The limited (<1000 m) effective dispersal observed for pollen and seeds is unexpected since the low weight of pollen and the good flying ability of poplar seeds. Long distance dispersal (LDD) of pollen and seeds may also be important in poplar, thought it does not seem to play a major role when looking at effective distances in current European habitats. However, artifacts due to the rarity of LDD events and the low number of seeds or seedlings analyzed cannot be excluded.
Statistical analysis on gene flow patterns
By the analysis of molecular data of seed and juvenile trees, Rathmacher et al. ([29]) could draw some general conclusions about distance and frequency of pollen and seed dispersal. Such data describe the actual dispersal events very precisely, but their use is limited by the sample size. In order to generalize explicit events, dispersal kernels were constructed. Such kernels are useful for a wide range of ecological studies like analyzing dispersal distances and inferring mating patterns ([7], [27], [21],[31]). In most cases, specific functions are fitted to the empirical data and conclusions are drawn on the basis of the best fitted function. The quality of these fits are varying with sample size. It is often the case that the data set contains many observations at short distances and only a few at larger distances. In all cases these fits entail a statistical uncertainty, whose level is dependent on the sample size and the unequal or skewed distribution of pairwise distances. Larger sample size lead to a low level of uncertainty of the shape of the fitted function, but at large distances only a few observations are often available, and therefore uncertainty increases. In the current literature this uncertainty is rarely considered.
Niggemann et al. ([26]) developed a new method using marked point processes for nonparametric estimation of dispersal kernels. This method allows to construct simulation envelopes of the given null model of random mating. These envelopes are a good representation of the uncertainty level of the derived dispersal kernel at a specific distance. In their paper, Niggemann et al. ([26]) applied this method to the dataset by Rathmacher et al. ([29]), consisting of genetic paternity data from an open pollinated P. nigra stand interspersed and surrounded by P. × canadensis trees. Statistical analysis showed a significant departure from the null model of random mating. Furthermore, calculating the mark correlation function for various distances and randomizing the marks allowed to obtain a simulation envelop for these data. The distances at which this function falls outside the simulation envelop indicate a significant departure from the null random mating model. Metadata analysis revealed a significant excess of mating events at short distances (up to 400 m) and a shortage of mating events at larger distances (1500-2000 m; Fig. 5 in [26]). Due to the low number of observations, the simulation envelopes became very wide at distances > 2000 m, indicating a large uncertainty in kernel estimation, and therefore results based on them should be taken with care. Comparison of these results with kernel fits using different maximum-likelihood models revealed a good match. From all tested models the two-component dispersal kernel was the best fitting function.
This approach of nonparametric kernel estimation should be used for data sets where sample size is limited and uncertainty of the measures is an important issue, as in the cases of risk analysis of transgenic outcrossing. Individual kernel fits might be similar to those obtained by classical approaches, but the level of uncertainty can not be derived from these methods. Finally, the results of the above analyzes clearly suggest the distance at which uncertainty becomes too large to draw any conclusions. This is always the case at larger distances due to sample size limitations.
Differences in germination rates
Next in the chain of monitoring trangene escape is the investigation of germination rates of seeds carrying artificial gene constructs in relation to seed from locally adapted P. nigra trees. Theoretical models already handled the higher or lower ecological advantages of transgenic species in the wild ([20]), but experimental data are not available on this topic.
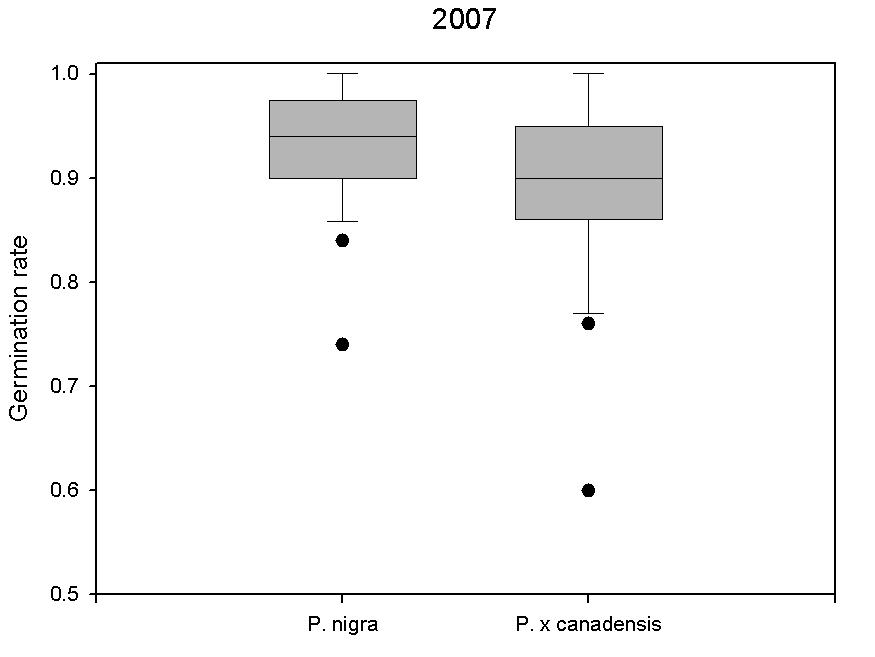
We studied the differences in germination rates between P. nigra and P. × canadensis ([19], [24]) by collecting seeds from 13 trees belonging to both taxa in the year 2007. In order to analyze the maternal effect, seeds were collected directly at the mother tree when the catkins burst started and wool squeezed out. Seeds were dried and the wool was removed using air pressure. Seeds were sown on filter paper in Petri dishes with Vermiculite, each dish containing 50 seeds from a single mother tree with four repetitions. Germination rates were determined after four days since sowing. Significant differences (p < 0.05) were found in germination rates between taxa, with P. nigra showing values slightly higher than P. × canadensis (93% vs. 89% - Fig. 2). This is well in accordance with data reported by Karrenberg & Suter ([18]), who found a viability rate of 91% for P. nigra. Both investigations were carried out under non-competing conditions. However, it is still not known if there are difference in their competition ability when both taxa compete for the same resources. Such studies are in progress ([19]). Findings of these investigations will be important to parametrize simulation models and enhance their prediction ability.
Fig. 2 - Germination rates of seeds from 15 different mother trees of the year 2007 differentiated by taxa (P. nigra and P. × canadensis).
Modeling transgene introgression rates
The use of simulation models in the assessment of transgenic outcrossing rates is important for several aspects of forest tree life-cycle hampering direct experimental research activities: (1) longevity of trees; (2) high amount of pollen produced; and (3) ability of long-distance pollen and seed dispersal. Furthermore, outcrossing rates needs to be estimated beforehand, in order to minimize the risk of outcrossing. Such models have been already developed ([11], [12], [20]) and several aspects dealing with transgene escape simulated. For example ([21]) investigated the rate of transgenic escape in cases where the modified organism carries mitigation genes, and DiFazio ([11]) used his model to predict transgenic escape assuming long distance dispersal as a common phenomenon. The model has been extensively parametrized using empirical data from aspen plantations in the United States.
All the above models are relatively complex and computer-intensive. A simpler and more efficient model based on the concept of cellular automata ([1]) has been developed within a joint research project dealing with transgenic woody species. Each cell represents a suitable habitat for the species and carries a whole population of trees with a certain population size. The carrying capacity of such a population is exactly the same for all habitat cells. All other cells are non-suitable habitat.
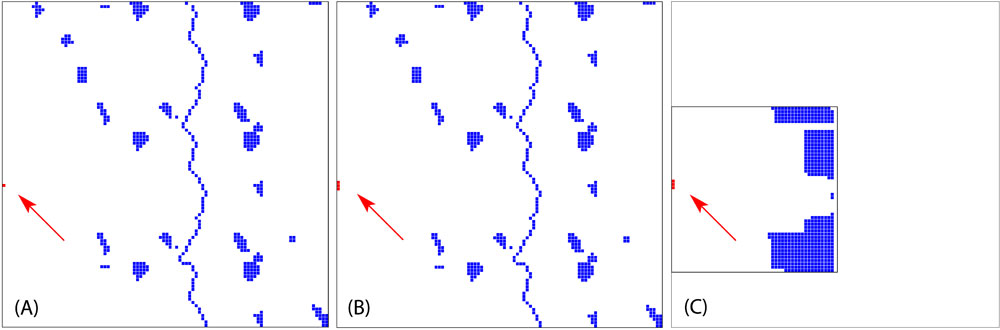
The aim of the project was to develop worst case scenarios on the usage of transgenic tree species in the landscape. Since no experimental data were available for both pollen and seed dispersal, as well as for effective pollination distances, a pure theoretical setup has been adopted ([4]). Within the virtual landscapes, we varied the size of transgenic populations (GMO) and the density of natural populations (Fig. 3). The total amount of natural populations has been kept constant in all landscapes. The fragmented landscape had been modeled after a typical northern Germany landscape. The vertical blue line in the center of diagrams displayed in Fig. 3a and Fig. 3b represents poplar populations along a river or a street.
Fig. 3 - The spatial setup of theoretical landscapes. All landscapes have the same amount of receiving trees (blue squares). The first two (A and B) show a fragmented landscape where receiving population are scattered. The third landscape (C) is a dense landscape. The first landscape (A) have a small transgenic plantation (red square), whereas the two others (B, C - indicated by the red arrows) have a plantation three times the size of the first one.
Simulations have been carried out using the three landscapes displayed in Fig. 3. The transgenic plantations were allowed to disperse their pollen for 50 years and then harvested. The amount of transgenic alleles released in the landscape was recorded for 100 independent simulations (Fig. 4). After harvesting of the GMO plantations, no more new genetic modified (GM) alleles were released into the landscapes: indeed, the amount of GM alleles remains roughly constant through time (Fig. 4a), as expected from the theoretical population model. All populations within the virtual landscapes are connected by gene flow. An increase of the GMO plantation size by the factor three increases also the frequency of transgenic escape by the factor three as expected (Fig. 4b). Keeping the GMO plantation size constant but setting the fragmentation of all natural populations to a minimum value, the frequency of transgenic escape is reduced back to the values with only one third of the GMO plantation (Fig. 4c). Even the maximum amount of GM alleles in the landscape is reduced back to the previous level (“Max” in Fig. 4).
Fig. 4 - Persistence of GMO alleles in the landscape after logging. The order of the landscapes is the same as in Fig. 2. (A) and (B) show the results of the fragmented landscape and (C) show the results of the dense landscape. In left diagram (A), the GMO plantation consists of only one cell, while in the central (B) and right (C) diagrams, the GMO plantation consists of three cells. The vertical line at the beginning (50 years) indicates the time the GMO plantation has been harvested. (count): frequency of transgenic escape; (Max): maximum amount of GM alleles in the landscape after 1000 simulated years.
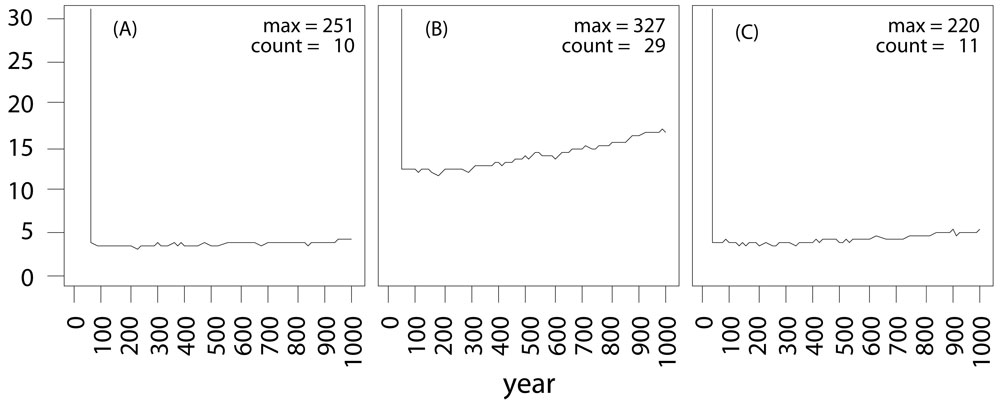
The importance of the GMO plantation size and its distance from potential recipients to control transgenic escape has been already reported ([17]). In addition, the simulations presented here highlights that density of the receiving population is an important parameter for the amount of outcrossing of GM alleles into the landscape. Such results can be obtained only within a reasonable time frame using theoretical simulation models.
Conclusion
This paper summarizes the results from three different research projects dealing with estimation and risk analysis of transgenic outcrossing in European landscapes. Although the aspects investigated do not cover the whole set of factors and processes responsible for transgenic outcrossing in plantations, the selected aspects are at the main focus of current risk analysis. This work dealt mainly with aspects of vertical gene transfer between GM plants and their native relatives. Because of the lack of experimental evidence on GM trees, the poplar complex of P. nigra as the native species and P. × canadensis as the potential GM plant was used as the model system. This is a very practical approach, since genetic modifications will concern only small parts of the genome, and therefore matings with the native relatives do not appear to be hampered. The use of taxa already planted in the landscape has the advantage that the environmental conditions like weather, wind and water are realistic, and no artificial corrections have to be made. One drawback of this approach may be that repetitions are only possible in the very same year at different locations, and changing climatic conditions are only possible by repeating the same experiments in different years.
The methods and results presented here are suitable for plantation forestry. Genetic modification of trees and their usage at large scale in forestry needs to be well prepared and evaluated. The long time span such organisms would remain in the natural environment and their potential to release transgenic pollen and seeds give to trees a high potential for transgene outcrossing. Even in short rotation plantations, an accidental release of transgenes in the environment may occur due to early flowering of single clones. Furthermore, short-rotation plantations involving willows (flowering at the age of 2-3 years) need to be carefully managed in order to get them into a profitable range.
Many attempts are made by scientists to prevent flowering and outcrossing of transgenic organisms. However, until such organisms will be availabile and tested, additional precautions to prevent accidental release of transgenes in the natural environment are needed. Simulations show that parameters such as distance and density of receiving populations may be a good starting point to minimize the risk of transgenic escape.
The main results from the reported research projects may be summarized as follows. Forest breeding programs need to evaluate the potentials of molecular techniques to provide fast growing clones for plantation forestry within a short timespan. For risk assessment it is important to look at all processes involved in transgene outcrossing. We found indications that the time of flowering itself may prevent outcrossing, since most planted poplar hybrids do flower earlier than the native P. nigra. For the time being, however, there are not enough data on the inheritance of flowering onset and duration on poplar hybrids. On the other hand, flowering timing may also be altered by applying genetic engineering methods. Another important aspect are the dispersal distances of pollen and seeds. In our study we found that the main amount of pollen (75%) and seeds (86%) are dispersed within distances < 1000 m. Based on the above evidence, dispersal kernels may be fitted for the use in simulation models, but care needs to be taken on the uncertainty of estimates mainly at larger distances, due to sample size limitations. Finally, simulation studies did reveal that the population size and the distance between donor and recipient population have a remarkable effect on the occurrence of outcrossing.
Acknowledgments
This work has been supported by the German Federal Environment Agency (FKZ: 201.67.4307/02) and by the German Federal Ministry of Education and Research (FKZ: 0313285J). I thanks all of my students and collaborators who help me in collecting and managing all the data presented in this work.
This work has been presented at the “First Biosafety Workshop” of COST Action FP0905, held in Hamburg (Germany) on September 9th, 2010.
References
Gscholar
Gscholar
Gscholar
Gscholar
Gscholar
Authors’ Info
Authors’ Affiliation
Dept. of Conservation Biology, Philipps University of Marburg, Karl-von-Frisch-Straβe 8, D-35032 Marburg (Germany)
Corresponding author
Paper Info
Citation
Bialozyt R (2012). Gene flow in poplar - experiments, analysis and modeling to prevent transgene outcrossing. iForest 5: 147-152. - doi: 10.3832/ifor0618-005
Academic Editor
Gabriele Bucci
Paper history
Received: Jan 24, 2012
Accepted: May 23, 2012
First online: Jun 13, 2012
Publication Date: Jun 29, 2012
Publication Time: 0.70 months
Copyright Information
© SISEF - The Italian Society of Silviculture and Forest Ecology 2012
Open Access
This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Web Metrics
Breakdown by View Type
Article Usage
Total Article Views: 56302
(from publication date up to now)
Breakdown by View Type
HTML Page Views: 46108
Abstract Page Views: 4031
PDF Downloads: 4400
Citation/Reference Downloads: 36
XML Downloads: 1727
Web Metrics
Days since publication: 5158
Overall contacts: 56302
Avg. contacts per week: 76.41
Article Citations
Article citations are based on data periodically collected from the Clarivate Web of Science web site
(last update: Mar 2025)
Total number of cites (since 2012): 9
Average cites per year: 0.64
Publication Metrics
by Dimensions ©
Articles citing this article
List of the papers citing this article based on CrossRef Cited-by.
Related Contents
iForest Similar Articles
Research Articles
Early flowering and genetic containment studies in transgenic poplar
vol. 5, pp. 138-146 (online: 13 June 2012)
Research Articles
Genetic variation and heritability estimates of Ulmus minor and Ulmus pumila hybrids for budburst, growth and tolerance to Ophiostoma novo-ulmi
vol. 8, pp. 422-430 (online: 15 December 2014)
Research Articles
Seedling emergence capacity and morphological traits are under strong genetic control in the resin tree Pinus oocarpa
vol. 17, pp. 245-251 (online: 16 August 2024)
Research Articles
Patterns of genetic variation in bud flushing of Abies alba populations
vol. 11, pp. 284-290 (online: 13 April 2018)
Research Articles
Genetic diversity of core vs. peripheral Norway spruce native populations at a local scale in Slovenia
vol. 11, pp. 104-110 (online: 31 January 2018)
Review Papers
Fifteen years of forest tree biosafety research in Germany
vol. 5, pp. 126-130 (online: 13 June 2012)
Research Articles
Age trends in genetic parameters for growth and quality traits in Abies alba
vol. 9, pp. 954-959 (online: 07 July 2016)
Review Papers
Genetic diversity and forest reproductive material - from seed source selection to planting
vol. 9, pp. 801-812 (online: 13 June 2016)
Research Articles
Comparison of genetic parameters between optimal and marginal populations of oriental sweet gum on adaptive traits
vol. 11, pp. 510-516 (online: 18 July 2018)
Research Articles
Fine-scale spatial genetic structure in a multi-oak-species (Quercus spp.) forest
vol. 8, pp. 324-332 (online: 05 September 2014)
iForest Database Search
Search By Author
Search By Keyword
Google Scholar Search
Citing Articles
Search By Author
Search By Keywords
PubMed Search
Search By Author
Search By Keyword






